Tilan Jason U, Lu Congyi, Galli Susana, Izycka-Swieszewska Ewa, Earnest Joshua Patrick, Shabbir Asim, Everhart Lindsay M, Wang Shuo, Martin Samantha, Horton Meredith, Mahajan Akanksha, Christian David, O'Neill Alison, Wang Hongkun, Zhuang Tingting, Czarnecka Magdalena, Johnson Michael D, Toretsky Jeffrey A, Kitlinska Joanna
Department of Nursing, Georgetown University Medical Center, Georgetown University, Washington DC.
Oncotarget. 2013 Dec;4(12):2487-501. doi: 10.18632/oncotarget.1604.
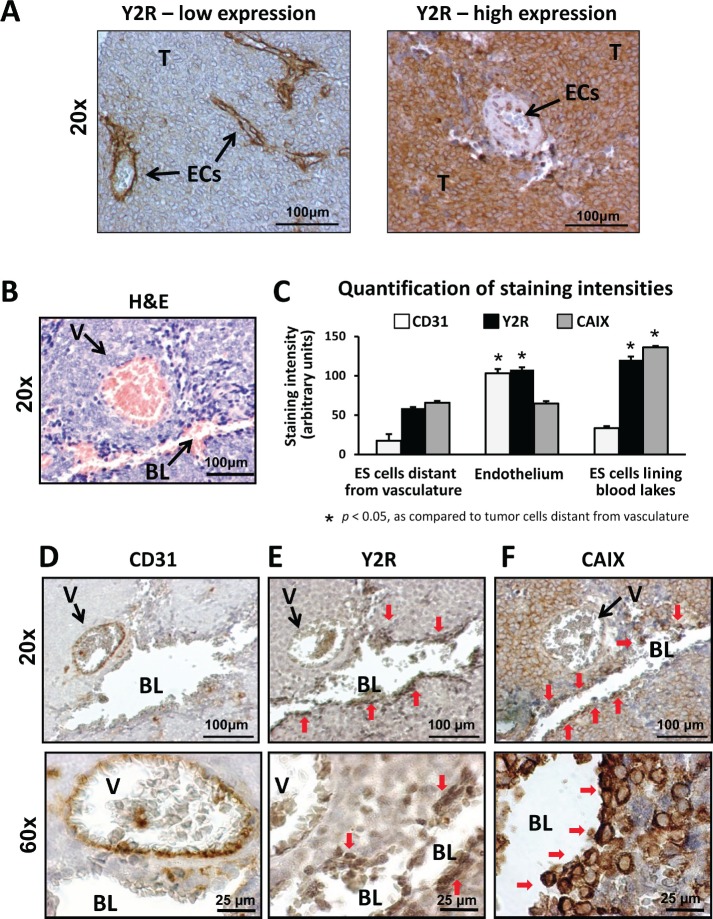
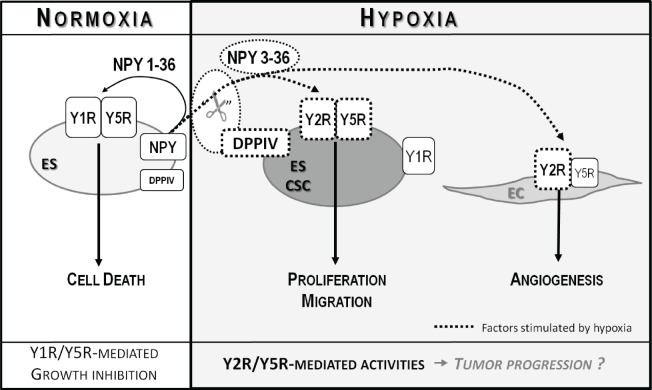
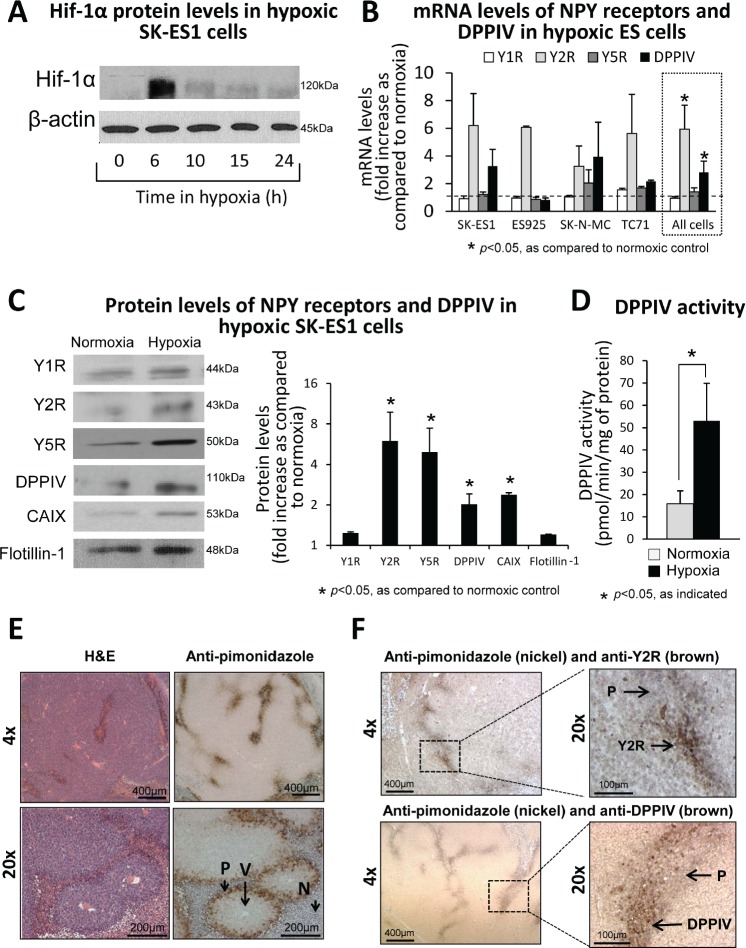
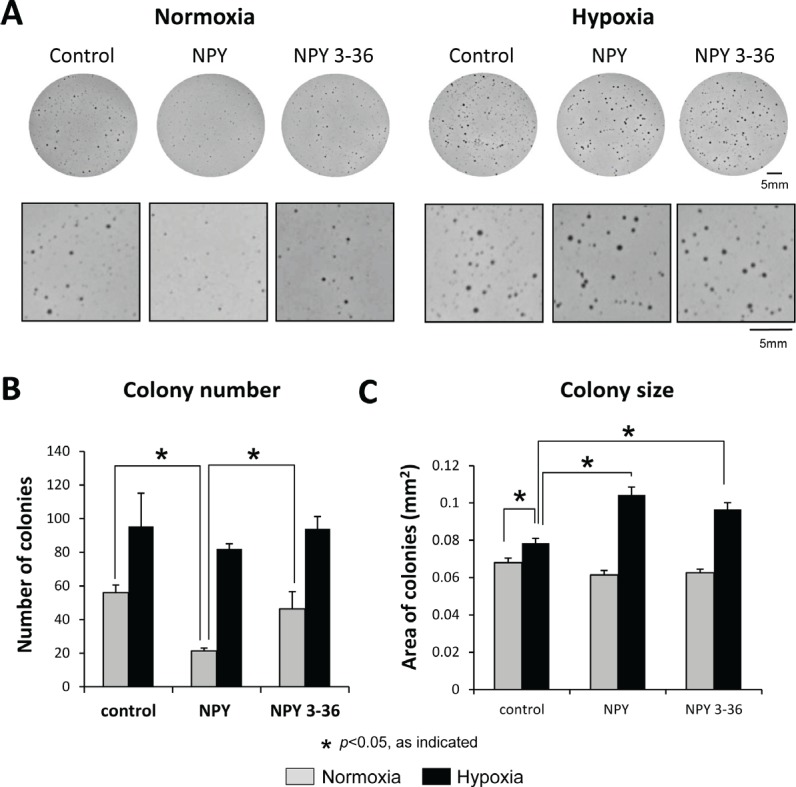
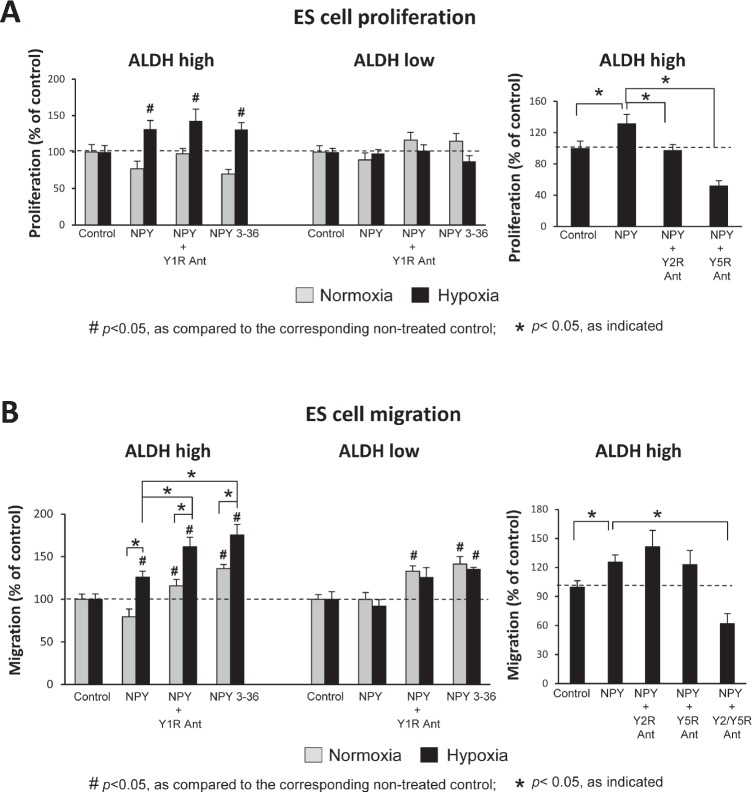
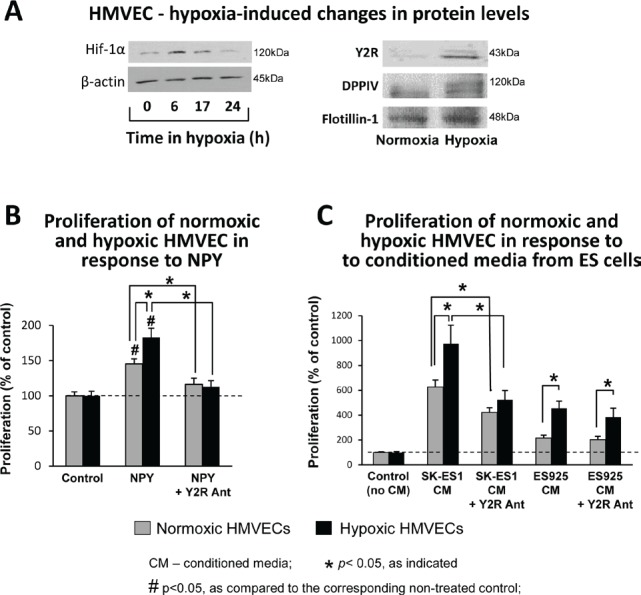
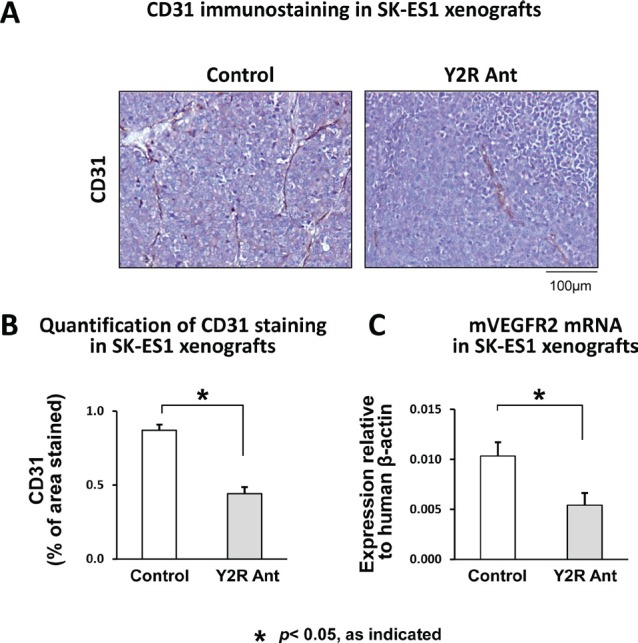
Ewing sarcoma (ES) is an aggressive malignancy driven by an oncogenic fusion protein, EWS-FLI1. Neuropeptide Y (NPY), and two of its receptors, Y1R and Y5R are up-regulated by EWS-FLI1 and abundantly expressed in ES cells. Paradoxically, NPY acting via Y1R and Y5R stimulates ES cell death. Here, we demonstrate that these growth-inhibitory actions of NPY are counteracted by hypoxia, which converts the peptide to a growth-promoting factor. In ES cells, hypoxia induces another NPY receptor, Y2R, and increases expression of dipeptidyl peptidase IV (DPPIV), an enzyme that cleaves NPY to a shorter form, NPY3-36. This truncated peptide no longer binds to Y1R and, therefore, does not stimulate ES cell death. Instead, NPY3-36 acts as a selective Y2R/Y5R agonist. The hypoxia-induced increase in DPPIV activity is most evident in a population of ES cells with high aldehyde dehydrogenase (ALDH) activity, rich in cancer stem cells (CSCs). Consequently, NPY, acting via Y2R/Y5Rs, preferentially stimulates proliferation and migration of hypoxic ALDHhigh cells. Hypoxia also enhances the angiogenic potential of ES by inducing Y2Rs in endothelial cells and increasing the release of its ligand, NPY3-36, from ES cells. In summary, hypoxia acts as a molecular switch shifting NPY activity away from Y1R/Y5R-mediated cell death and activating the Y2R/Y5R/DPPIV/NPY3-36 axis, which stimulates ES CSCs and promotes angiogenesis. Hypoxia-driven actions of the peptide such as these may contribute to ES progression. Due to the receptor-specific and multifaceted nature of NPY actions, these findings may inform novel therapeutic approaches to ES.
尤因肉瘤(ES)是一种由致癌融合蛋白EWS-FLI1驱动的侵袭性恶性肿瘤。神经肽Y(NPY)及其两个受体Y1R和Y5R在EWS-FLI1的作用下上调,并在ES细胞中大量表达。矛盾的是,通过Y1R和Y5R起作用的NPY会刺激ES细胞死亡。在此,我们证明NPY的这些生长抑制作用被缺氧所抵消,缺氧将该肽转化为一种生长促进因子。在ES细胞中,缺氧诱导另一种NPY受体Y2R,并增加二肽基肽酶IV(DPPIV)的表达,DPPIV是一种将NPY切割成较短形式NPY3-36的酶。这种截短的肽不再与Y1R结合,因此不会刺激ES细胞死亡。相反,NPY3-36作为一种选择性Y2R/Y5R激动剂。缺氧诱导的DPPIV活性增加在具有高醛脱氢酶(ALDH)活性、富含癌症干细胞(CSC)的ES细胞群体中最为明显。因此,通过Y2R/Y5R起作用的NPY优先刺激缺氧的ALDH高表达细胞的增殖和迁移。缺氧还通过在内皮细胞中诱导Y2R并增加其配体NPY3-36从ES细胞中的释放来增强ES的血管生成潜力。总之,缺氧作为一种分子开关,将NPY的活性从Y1R/Y5R介导的细胞死亡转变为激活Y2R/Y5R/DPPIV/NPY3-36轴,后者刺激ES CSC并促进血管生成。肽的这些缺氧驱动的作用可能有助于ES的进展。由于NPY作用的受体特异性和多方面性质,这些发现可能为ES的新型治疗方法提供思路。