Centre for Rare Diseases and Personalised Medicine, School of Clinical and Experimental Medicine, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK.
Clin Epigenetics. 2013 Dec 10;5(1):23. doi: 10.1186/1868-7083-5-23.
Beckwith-Wiedemann syndrome (BWS) is a congenital overgrowth disorder associated with abnormalities in 11p15.5 imprinted genes. The most common cause is loss of methylation (epimutation) at the imprinting control centre 2 (IC2/KvDMR1). Most IC2 epimutations occur sporadically but an association with conception after assisted reproductive technologies (ART) has been reported. A subgroup of IC2 epimutation cases also harbour epimutations at other imprinting centres (ICs) outside of 11p15.5. We have investigated the relationship between these multiple epimutation cases (ME+), history of ART and clinical phenotype in a cohort of 187 BWS IC2 epimutation patients.
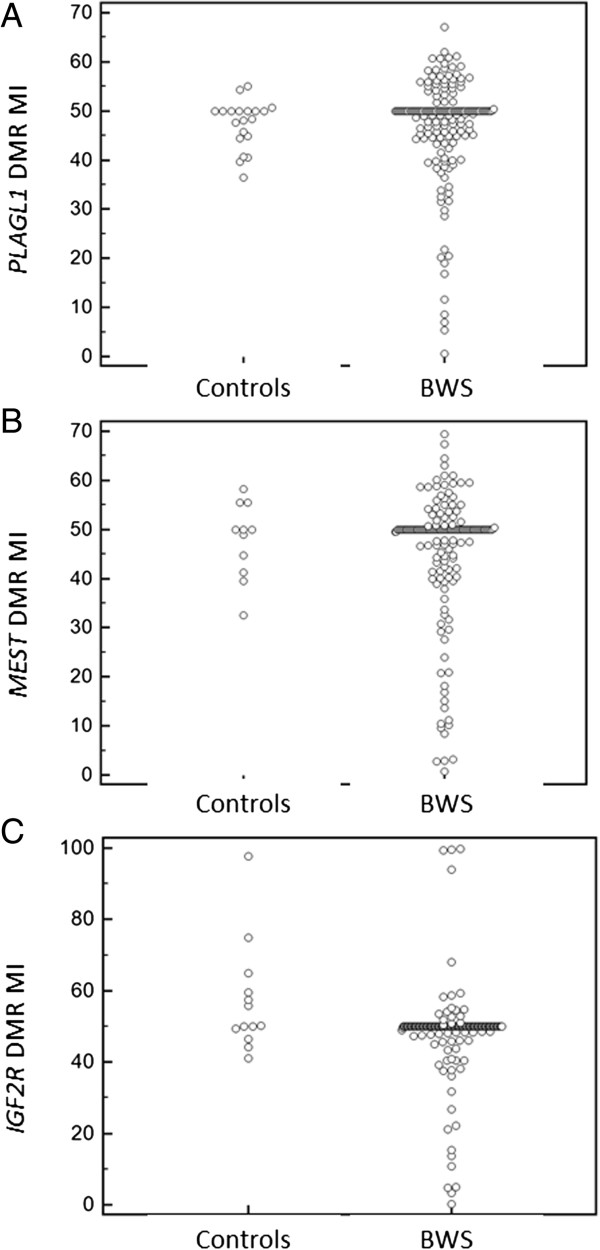
Methylation analysis at PLAGL1, MEST and IGF2R ICs demonstrated an over-representation of patients with abnormally low methylation (8.5%, 12% and 6% respectively). At IGF2R some patients (2%) had gain of methylation but this was also detected in controls. Though there were no significant correlations between the methylation index (MIs) at the three ICs tested, a subset of patients appeared to be susceptible to multiple epimutations (ME+) and 21.2% of ME + patients had been conceived by ART compared to 4.5% (P = 0.0033) without additional epimutations. Methylation array profiling (Illumina Goldengate®) of patients and controls (excluding 11p15.5 loci) demonstrated significant differences between patients and controls. No significant associations were found between aspects of the BWS phenotype and individual epimutations but we describe a case presenting with a post-ART BWS-like phenotype in which molecular analysis demonstrated loss of paternal allele methylation at the 11p15.5 IC1 locus (IC1 regulates imprinting of IGF2 and H19). Loss of paternal allele methylation at the IC1 is the molecular finding associated with Silver-Russell syndrome whereas BWS is associated with gain of maternal allele methylation at IC1. Further analysis demonstrated epimutations at PLAGL1 and MEST consistent with the hypothesis that the presence of multiple epimutations may be of clinical relevance.
These findings suggest that the ME + subgroup of BWS patients are preferentially, but not exclusively, associated with a history of ART and that, though at present, there are no clear epigenotype-phenotype correlations for ME + BWS patients, non-11p15.5 IC epimutations can influence clinical phenotype.
贝克威思-威德曼综合征(BWS)是一种与 11p15.5 印迹基因异常相关的先天性过度生长障碍。最常见的原因是印迹控制中心 2(IC2/KvDMR1)的甲基化缺失(表观遗传突变)。大多数 IC2 表观遗传突变是偶然发生的,但已有报道称其与辅助生殖技术(ART)后的受孕有关。亚组的 IC2 表观遗传突变病例也在 11p15.5 以外的其他印迹中心(IC)中存在表观遗传突变。我们在 187 名 BWS IC2 表观遗传突变患者的队列中研究了这些多表观遗传突变病例(ME+)、ART 史和临床表型之间的关系。
PLAGL1、MEST 和 IGF2R IC 的甲基化分析显示,异常低甲基化的患者比例较高(分别为 8.5%、12%和 6%)。在 IGF2R 中,一些患者(2%)存在甲基化增加,但在对照组中也检测到了这种情况。尽管测试的三个 IC 中的甲基化指数(MI)之间没有显著相关性,但似乎有一部分患者容易发生多表观遗传突变(ME+),21.2%的 ME+患者是通过 ART 受孕的,而没有其他表观遗传突变的患者为 4.5%(P=0.0033)。对患者和对照(不包括 11p15.5 基因座)进行的甲基化阵列分析(Illumina Goldengate®)显示,患者和对照之间存在显著差异。但我们没有发现 BWS 表型的各个方面与单个表观遗传突变之间存在显著关联,但我们描述了一个病例,该病例表现为 ART 后出现的 BWS 样表型,分子分析显示 11p15.5 IC1 位点(IC1 调节 IGF2 和 H19 的印迹)的父本等位基因甲基化缺失。IC1 处父本等位基因甲基化缺失是与银-罗素综合征相关的分子发现,而 BWS 与 IC1 处母本等位基因甲基化增加相关。进一步的分析显示 PLAGL1 和 MEST 存在表观遗传突变,这与多表观遗传突变可能具有临床相关性的假设一致。
这些发现表明,BWS 患者的 ME+亚组更倾向于(但并非排他性地)与 ART 史相关,而且,尽管目前 ME+BWS 患者的表型-表型关联尚无明确的表观遗传证据,但非 11p15.5 IC 表观遗传突变可影响临床表型。