Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, United States of America.
Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John's University, Queens, New York, United States of America.
PLoS One. 2013 Dec 5;8(12):e82463. doi: 10.1371/journal.pone.0082463. eCollection 2013.
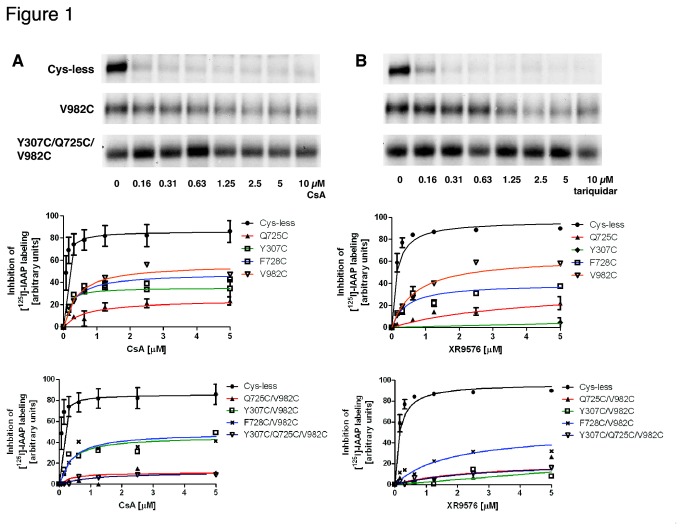
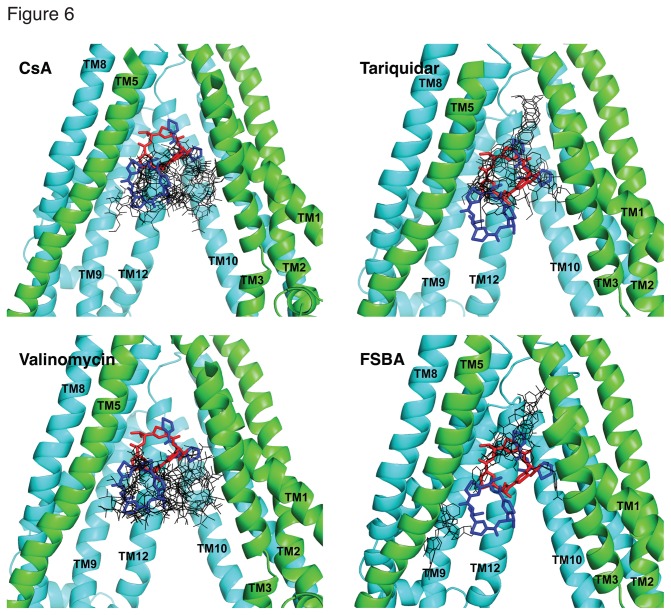
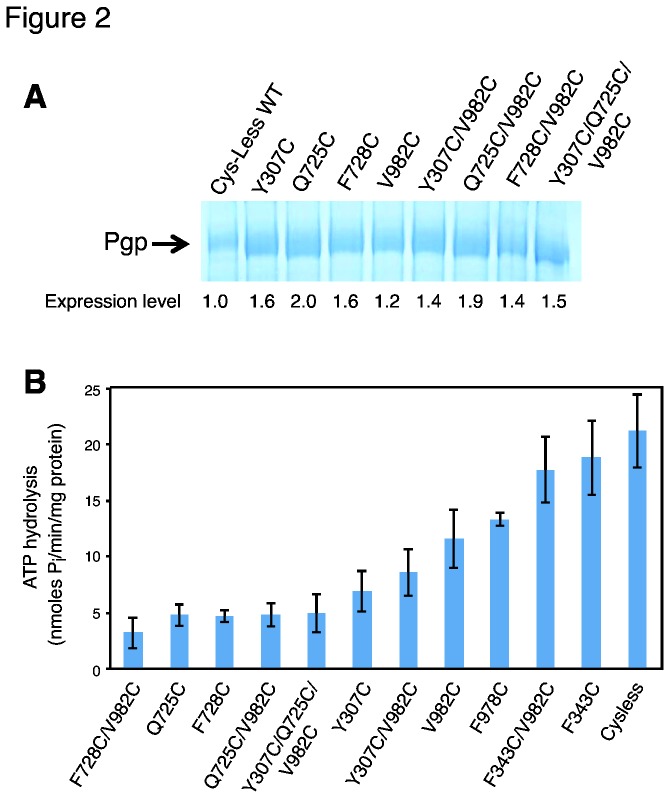
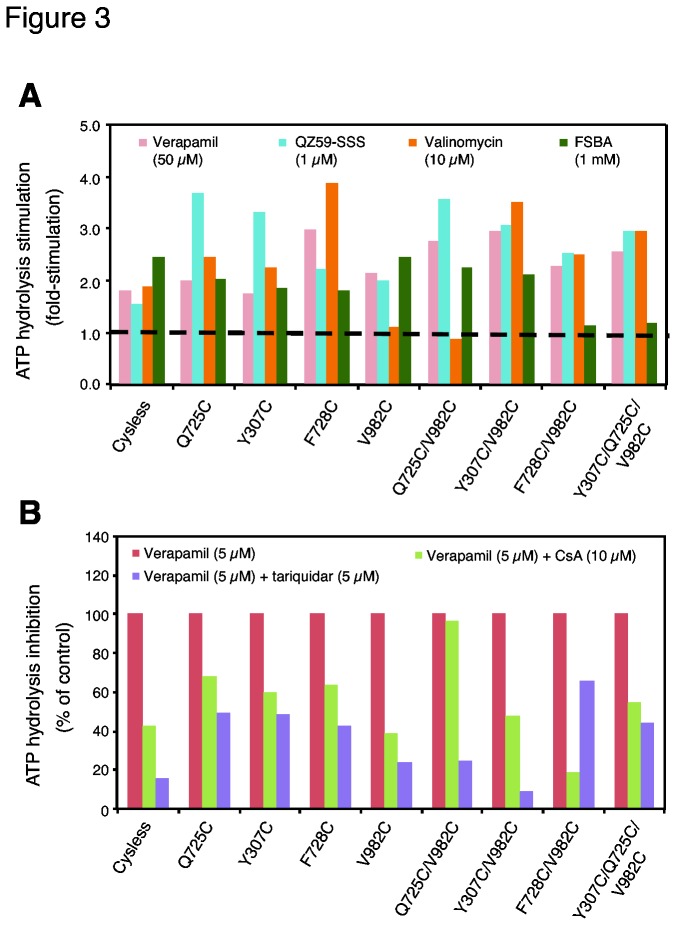
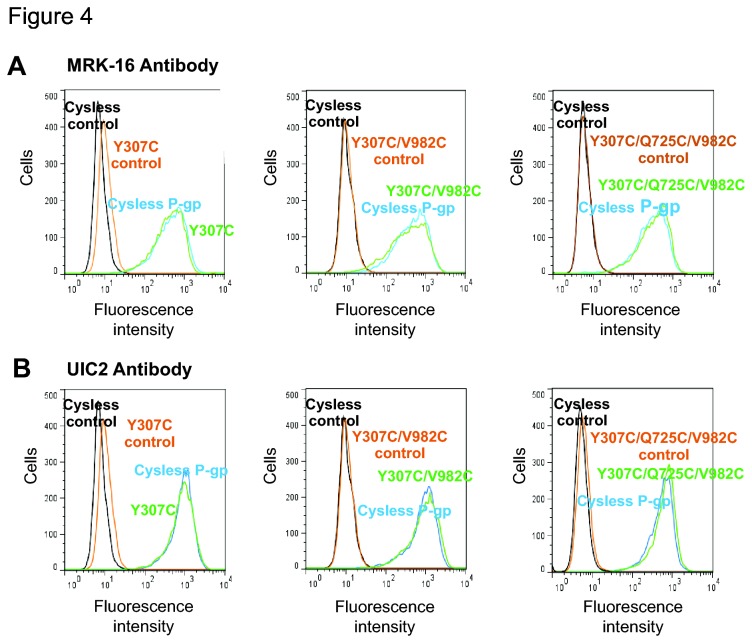
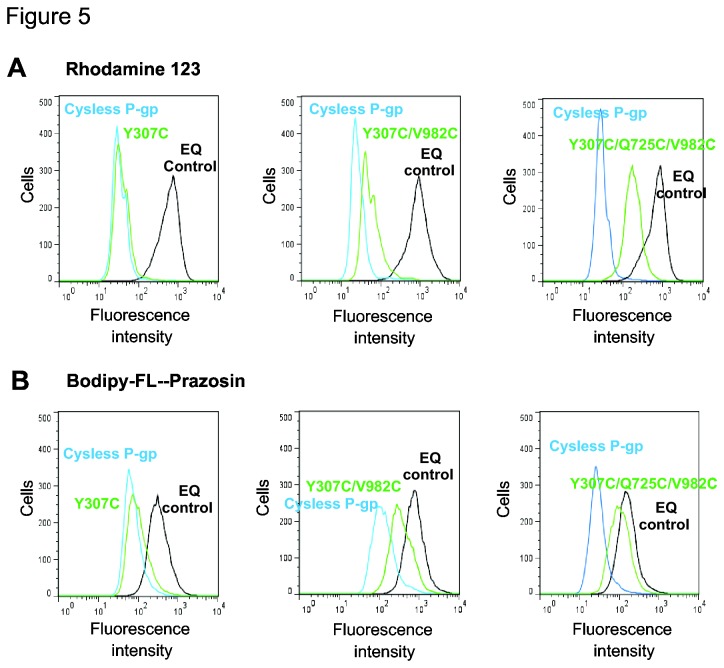
P-glycoprotein (Pgp, ABCB1) is an ATP-Binding Cassette (ABC) transporter that is associated with the development of multidrug resistance in cancer cells. Pgp transports a variety of chemically dissimilar amphipathic compounds using the energy from ATP hydrolysis. In the present study, to elucidate the binding sites on Pgp for substrates and modulators, we employed site-directed mutagenesis, cell- and membrane-based assays, molecular modeling and docking. We generated single, double and triple mutants with substitutions of the Y307, F343, Q725, F728, F978 and V982 residues at the proposed drug-binding site with cys in a cysless Pgp, and expressed them in insect and mammalian cells using a baculovirus expression system. All the mutant proteins were expressed at the cell surface to the same extent as the cysless wild-type Pgp. With substitution of three residues of the pocket (Y307, Q725 and V982) with cysteine in a cysless Pgp, QZ59S-SSS, cyclosporine A, tariquidar, valinomycin and FSBA lose the ability to inhibit the labeling of Pgp with a transport substrate, [(125)I]-Iodoarylazidoprazosin, indicating these drugs cannot bind at their primary binding sites. However, the drugs can modulate the ATP hydrolysis of the mutant Pgps, demonstrating that they bind at secondary sites. In addition, the transport of six fluorescent substrates in HeLa cells expressing triple mutant (Y307C/Q725C/V982C) Pgp is also not significantly altered, showing that substrates bound at secondary sites are still transported. The homology modeling of human Pgp and substrate and modulator docking studies support the biochemical and transport data. In aggregate, our results demonstrate that a large flexible pocket in the Pgp transmembrane domains is able to bind chemically diverse compounds. When residues of the primary drug-binding site are mutated, substrates and modulators bind to secondary sites on the transporter and more than one transport-active binding site is available for each substrate.
P-糖蛋白(Pgp,ABCB1)是一种与癌细胞多药耐药性发展相关的 ATP 结合盒(ABC)转运蛋白。Pgp 利用 ATP 水解产生的能量转运各种化学性质不同的两亲化合物。在本研究中,为了阐明 Pgp 与底物和调节剂结合的结合部位,我们采用了定点突变、细胞和膜基测定、分子建模和对接。我们生成了单个、双个和三个突变体,这些突变体取代了拟药物结合部位的 Y307、F343、Q725、F728、F978 和 V982 残基,并在无半胱氨酸的 Pgp 中用半胱氨酸取代,然后使用杆状病毒表达系统在昆虫和哺乳动物细胞中表达。所有突变蛋白在细胞表面的表达水平与无半胱氨酸的野生型 Pgp 相同。用无半胱氨酸的 Pgp 中三个口袋残基(Y307、Q725 和 V982)的半胱氨酸取代,QZ59S-SSS、环孢菌素 A、他利喹达、缬氨霉素和 FSBA 丧失抑制用转运底物[(125)I]-碘代氮杂泊司琼标记 Pgp 的能力,表明这些药物不能与其主要结合部位结合。然而,这些药物可以调节突变体 Pgps 的 ATP 水解,表明它们与次要部位结合。此外,在表达三重突变体(Y307C/Q725C/V982C)Pgp 的 HeLa 细胞中,六种荧光底物的转运也没有明显改变,表明结合在次要部位的底物仍被转运。人 Pgp 的同源建模和底物与调节剂对接研究支持了生化和转运数据。总之,我们的结果表明,Pgp 跨膜结构域中的一个大的柔性口袋能够结合化学性质不同的化合物。当主要药物结合部位的残基发生突变时,底物和调节剂结合到转运蛋白的次要部位,每个底物都有多个转运活性的结合部位。