Department of Biology, Molecular Biology and Heart Institutes, San Diego State University, San Diego, California, United States of America ; Development and Aging Program, Sanford-Burnham Institute for Medical Research, La Jolla, California, United States of America.
Department of Biology, Molecular Biology and Heart Institutes, San Diego State University, San Diego, California, United States of America.
PLoS Genet. 2013;9(12):e1004024. doi: 10.1371/journal.pgen.1004024. Epub 2013 Dec 19.
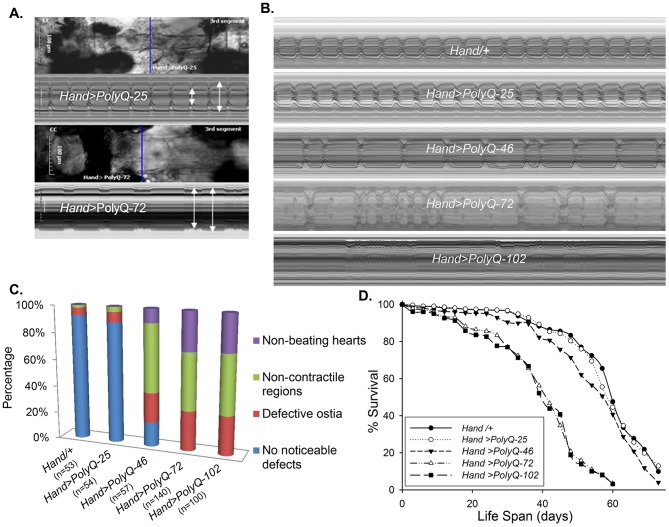
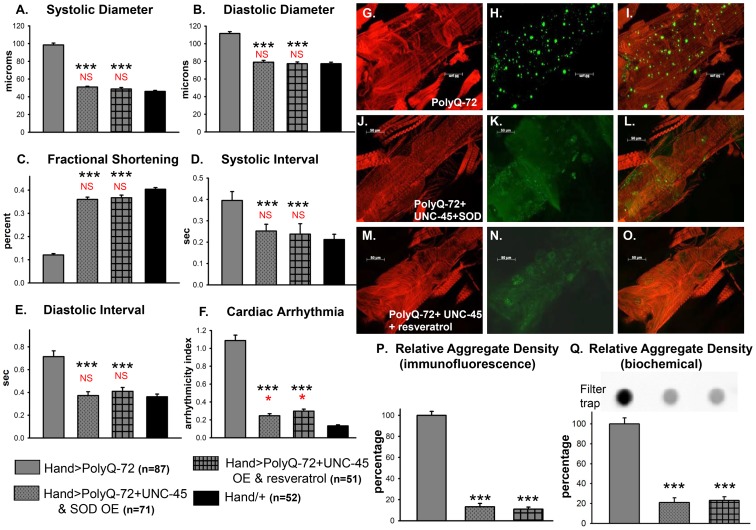
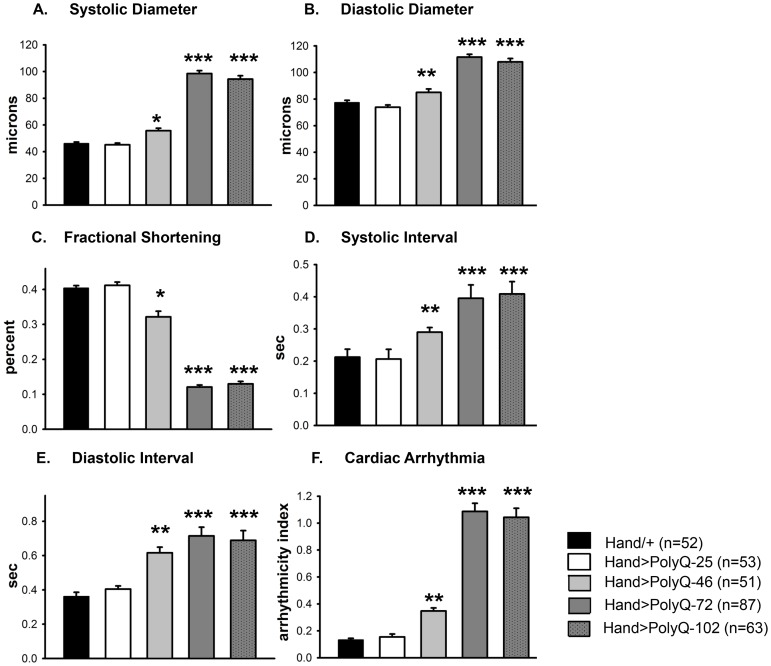
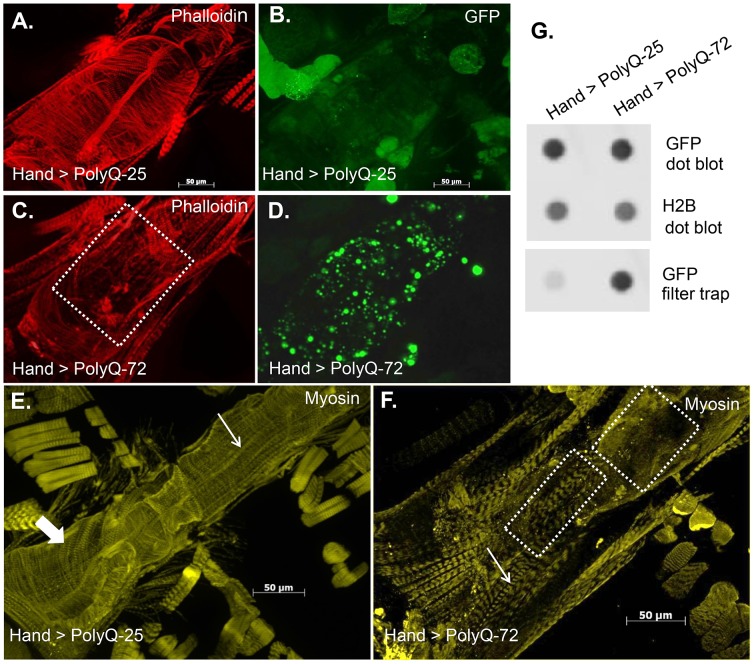
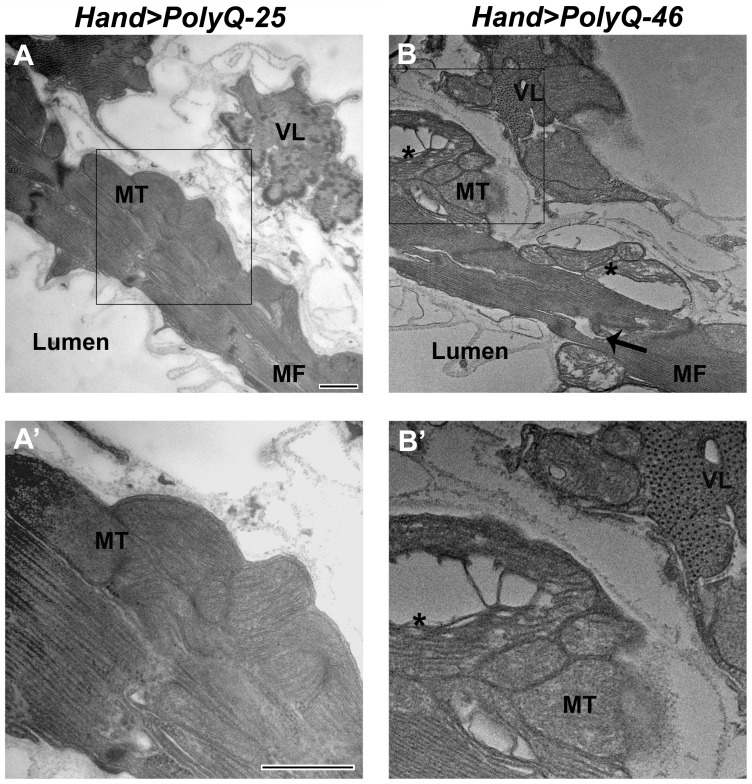
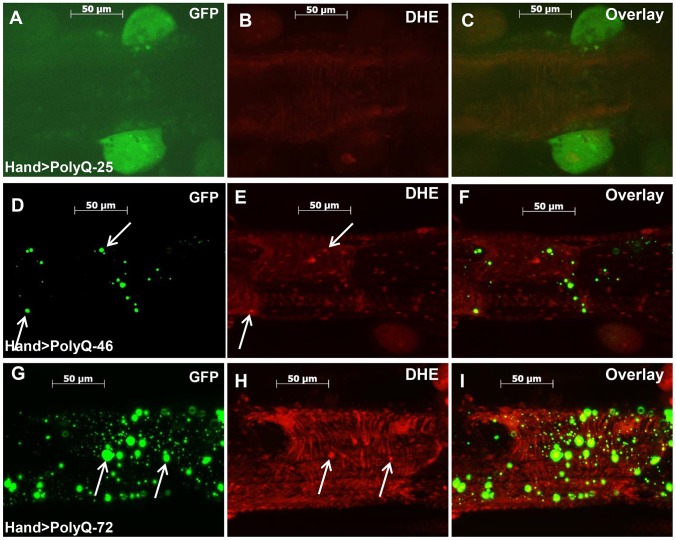
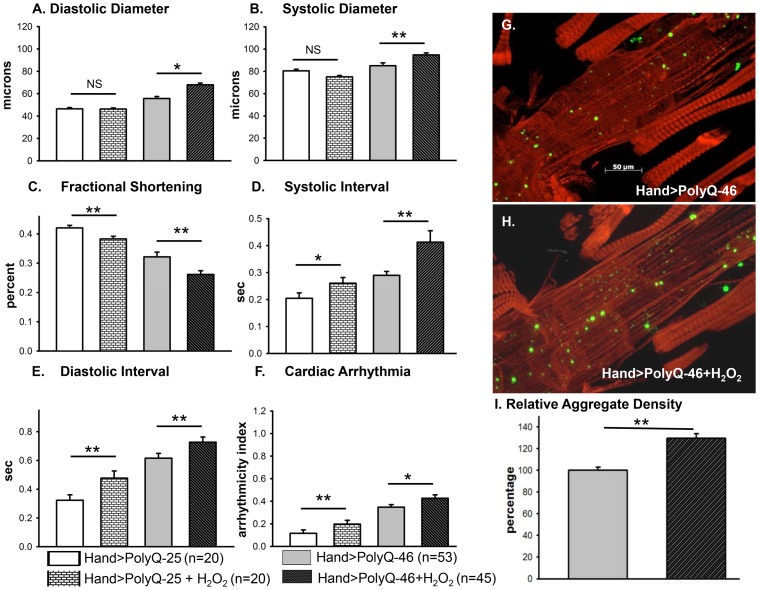
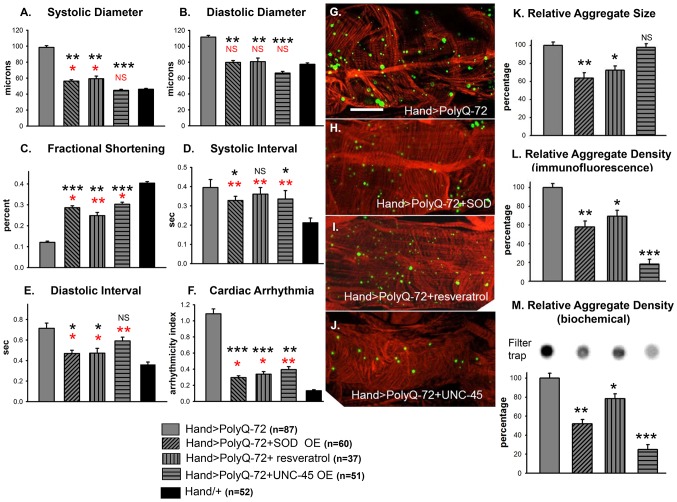
Amyloid-like inclusions have been associated with Huntington's disease (HD), which is caused by expanded polyglutamine repeats in the Huntingtin protein. HD patients exhibit a high incidence of cardiovascular events, presumably as a result of accumulation of toxic amyloid-like inclusions. We have generated a Drosophila model of cardiac amyloidosis that exhibits accumulation of PolyQ aggregates and oxidative stress in myocardial cells, upon heart-specific expression of Huntingtin protein fragments (Htt-PolyQ) with disease-causing poly-glutamine repeats (PolyQ-46, PolyQ-72, and PolyQ-102). Cardiac expression of GFP-tagged Htt-PolyQs resulted in PolyQ length-dependent functional defects that included increased incidence of arrhythmias and extreme cardiac dilation, accompanied by a significant decrease in contractility. Structural and ultrastructural analysis of the myocardial cells revealed reduced myofibrillar content, myofibrillar disorganization, mitochondrial defects and the presence of PolyQ-GFP positive aggregates. Cardiac-specific expression of disease causing Poly-Q also shortens lifespan of flies dramatically. To further confirm the involvement of oxidative stress or protein unfolding and to understand the mechanism of PolyQ induced cardiomyopathy, we co-expressed expanded PolyQ-72 with the antioxidant superoxide dismutase (SOD) or the myosin chaperone UNC-45. Co-expression of SOD suppressed PolyQ-72 induced mitochondrial defects and partially suppressed aggregation as well as myofibrillar disorganization. However, co-expression of UNC-45 dramatically suppressed PolyQ-72 induced aggregation and partially suppressed myofibrillar disorganization. Moreover, co-expression of both UNC-45 and SOD more efficiently suppressed GFP-positive aggregates, myofibrillar disorganization and physiological cardiac defects induced by PolyQ-72 than did either treatment alone. Our results demonstrate that mutant-PolyQ induces aggregates, disrupts the sarcomeric organization of contractile proteins, leads to mitochondrial dysfunction and increases oxidative stress in cardiomyocytes leading to abnormal cardiac function. We conclude that modulation of both protein unfolding and oxidative stress pathways in the Drosophila heart model can ameliorate the detrimental PolyQ effects, thus providing unique insights into the genetic mechanisms underlying amyloid-induced cardiac failure in HD patients.
淀粉样蛋白样包涵体与亨廷顿病(HD)有关,HD 是由亨廷顿蛋白中扩展的多聚谷氨酰胺重复引起的。HD 患者表现出心血管事件的高发率,推测是由于毒性淀粉样蛋白样包涵体的积累。我们已经生成了一种果蝇心脏淀粉样变性模型,该模型在心特异性表达具有致病多聚谷氨酰胺重复(PolyQ-46、PolyQ-72 和 PolyQ-102)的亨廷顿蛋白片段(Htt-PolyQ)时,表现出多聚 Q 聚集物和心肌细胞中氧化应激的积累。GFP 标记的 Htt-PolyQ 的心脏表达导致了与多聚 Q 长度相关的功能缺陷,包括心律失常发生率增加和心脏极度扩张,同时收缩性显著降低。心肌细胞的结构和超微结构分析显示肌原纤维含量减少、肌原纤维排列紊乱、线粒体缺陷和 PolyQ-GFP 阳性聚集物的存在。致病 Poly-Q 的心脏特异性表达也显著缩短了果蝇的寿命。为了进一步证实氧化应激或蛋白质展开的参与,并了解 PolyQ 诱导的心肌病的机制,我们共表达了扩展的 PolyQ-72 与抗氧化超氧化物歧化酶(SOD)或肌球蛋白伴侣 UNC-45。SOD 的共表达抑制了 PolyQ-72 诱导的线粒体缺陷,并部分抑制了聚集以及肌原纤维排列紊乱。然而,UNC-45 的共表达显著抑制了 PolyQ-72 诱导的聚集,并部分抑制了肌原纤维排列紊乱。此外,与单独治疗相比,UNC-45 和 SOD 的共表达更有效地抑制了 PolyQ-72 诱导的 GFP 阳性聚集物、肌原纤维排列紊乱和生理心脏缺陷。我们的结果表明,突变型-PolyQ 诱导聚集物、破坏收缩蛋白的肌节组织、导致线粒体功能障碍并增加心肌细胞中的氧化应激,导致异常的心脏功能。我们得出结论,在果蝇心脏模型中调节蛋白质展开和氧化应激途径可以改善有害的 PolyQ 效应,从而为 HD 患者淀粉样蛋白诱导的心力衰竭的遗传机制提供独特的见解。