Salzberg Steven L, Pertea Mihaela, Fahrner Jill A, Sobreira Nara
Center for Computational Biology, Johns Hopkins School of Medicine, Baltimore, Maryland, 21205; McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins School of Medicine, Baltimore, Maryland, 21205.
Hum Mutat. 2014 Mar;35(3):283-8. doi: 10.1002/humu.22503.
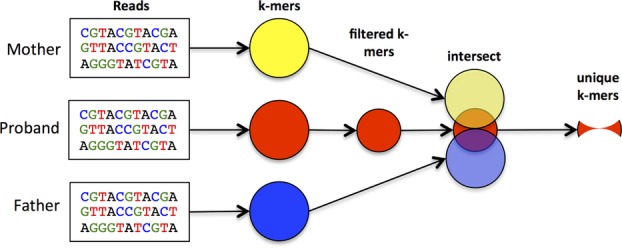
DNA sequencing has become a powerful method to discover the genetic basis of disease. Standard, widely used protocols for analysis usually begin by comparing each individual to the human reference genome. When applied to a set of related individuals, this approach reveals millions of differences, most of which are shared among the individuals and unrelated to the disease being investigated. We have developed a novel algorithm for variant detection, one that compares DNA sequences directly to one another, without aligning them to the reference genome. When used to find de novo mutations in exome sequences from family trios, or to compare normal and diseased samples from the same individual, the new method, direct alignment for mutation discovery (DIAMUND), produces a dramatically smaller list of candidate mutations than previous methods, without losing sensitivity to detect the true cause of a genetic disease. We demonstrate our results on several example cases, including two family trios in which it correctly found the disease-causing variant while excluding thousands of harmless variants that standard methods had identified.
DNA测序已成为发现疾病遗传基础的有力方法。标准的、广泛使用的分析方案通常首先将每个个体与人类参考基因组进行比较。当应用于一组相关个体时,这种方法会揭示数百万个差异,其中大多数差异在个体之间是共享的,并且与所研究的疾病无关。我们开发了一种用于变异检测的新算法,该算法直接将DNA序列相互比较,而无需将它们与参考基因组比对。当用于在三联体家庭的外显子组序列中寻找新生突变,或比较同一个体的正常样本和患病样本时,这种新方法——用于突变发现的直接比对(DIAMUND),产生的候选突变列表比以前的方法要小得多,同时又不会失去检测遗传病真正病因的敏感性。我们在几个示例案例中展示了我们的结果,包括两个三联体家庭,在这些案例中,它正确地找到了致病变异,同时排除了标准方法所识别的数千个无害变异。