Department of Molecular Biology, Max Planck Institute for Developmental Biology, Tübingen, Germany.
Nat Genet. 2020 May;52(5):534-540. doi: 10.1038/s41588-020-0612-7. Epub 2020 Apr 13.
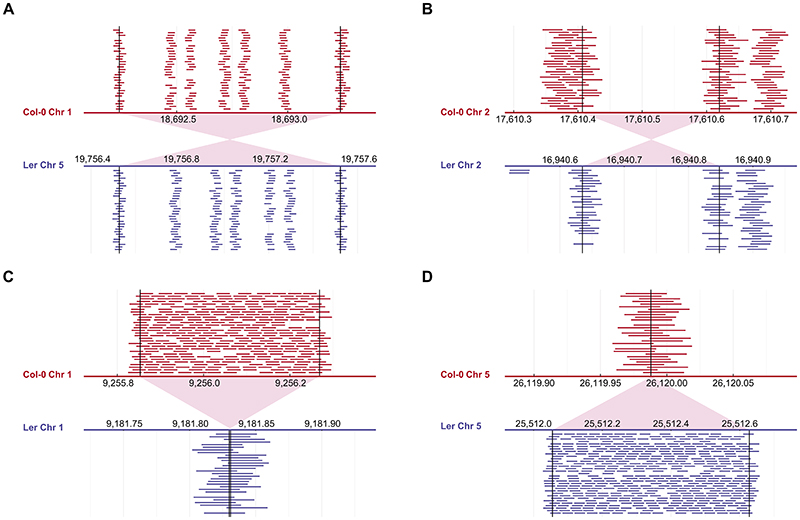
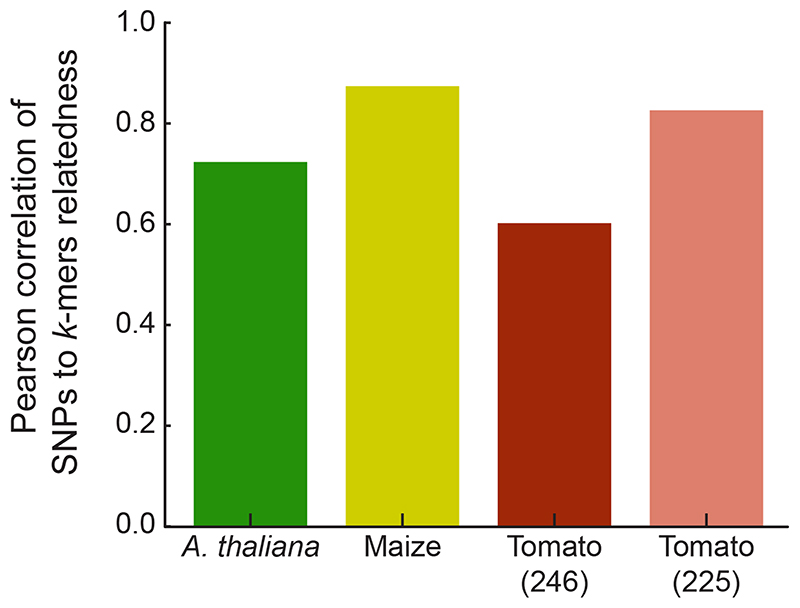
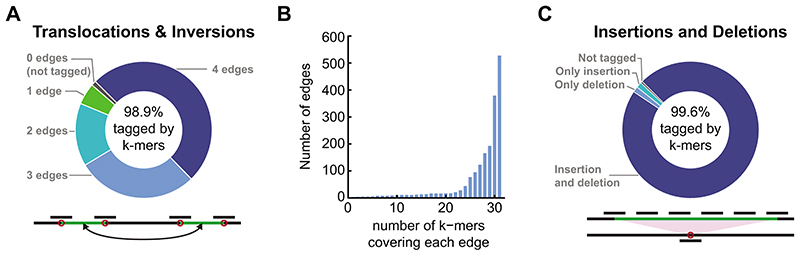
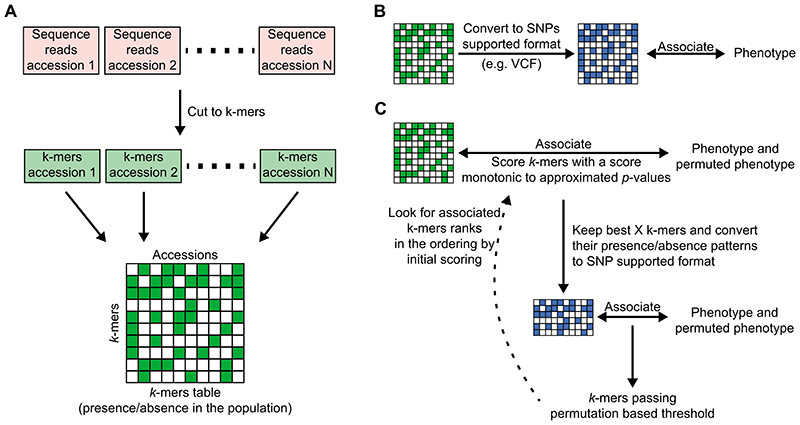
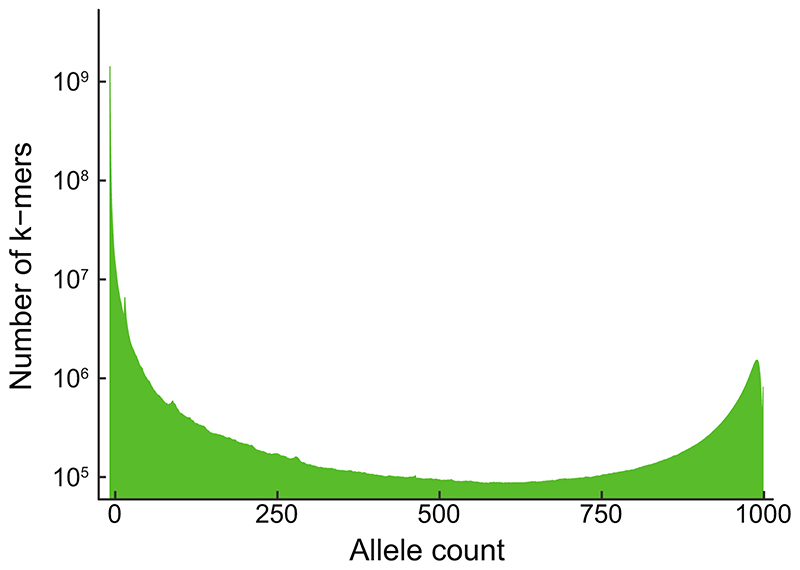
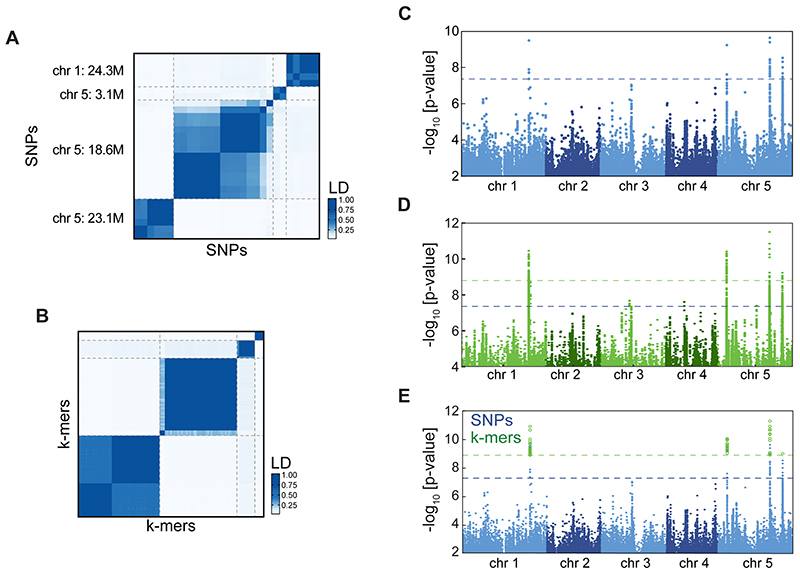
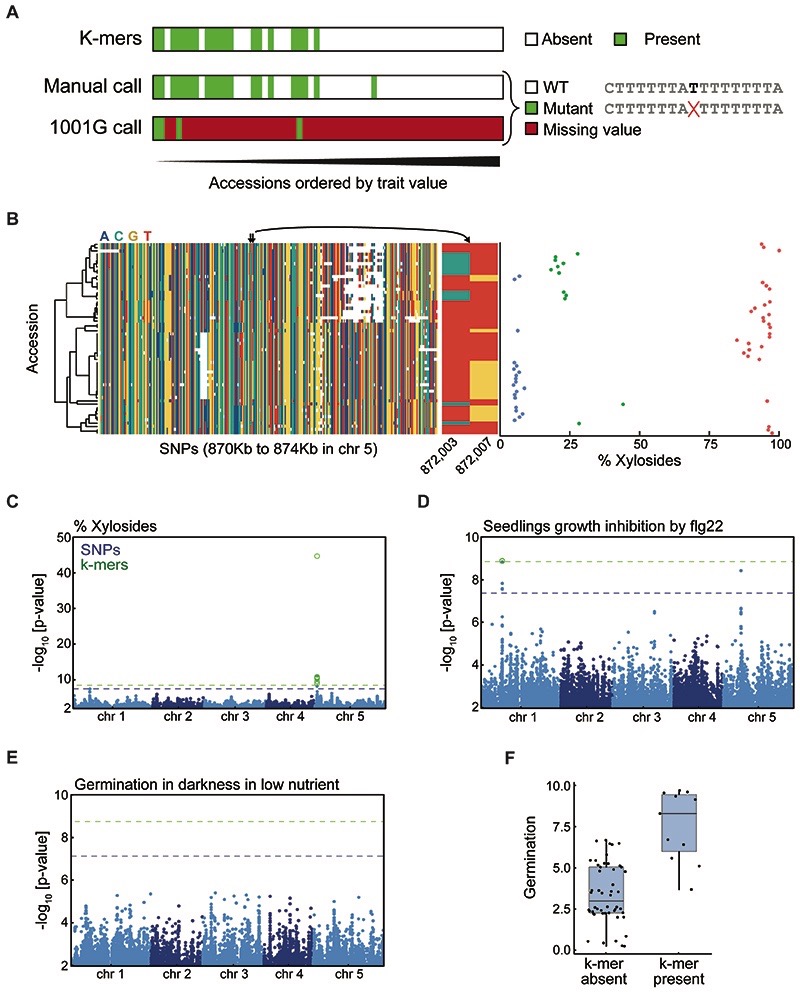
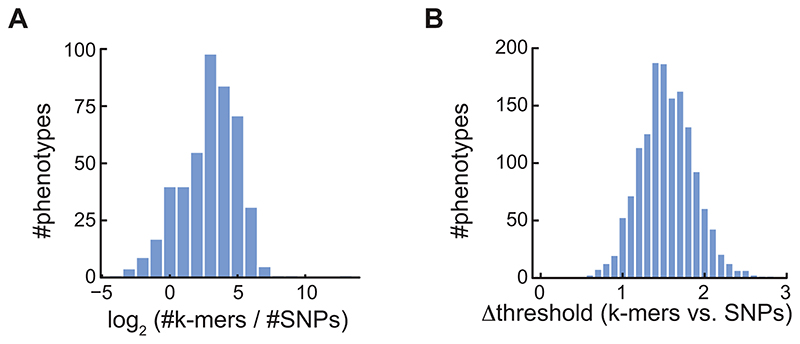
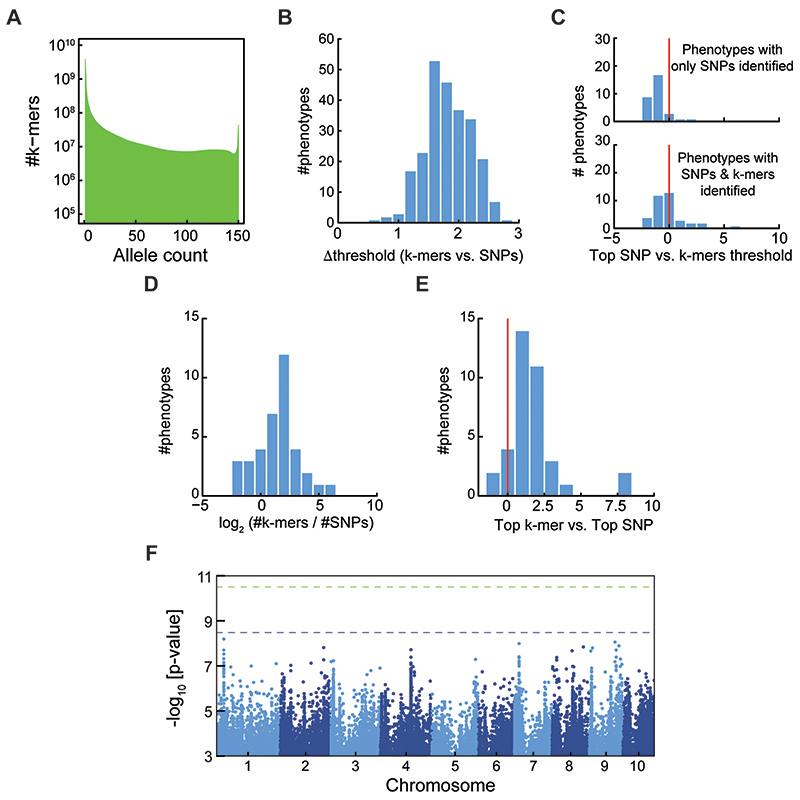
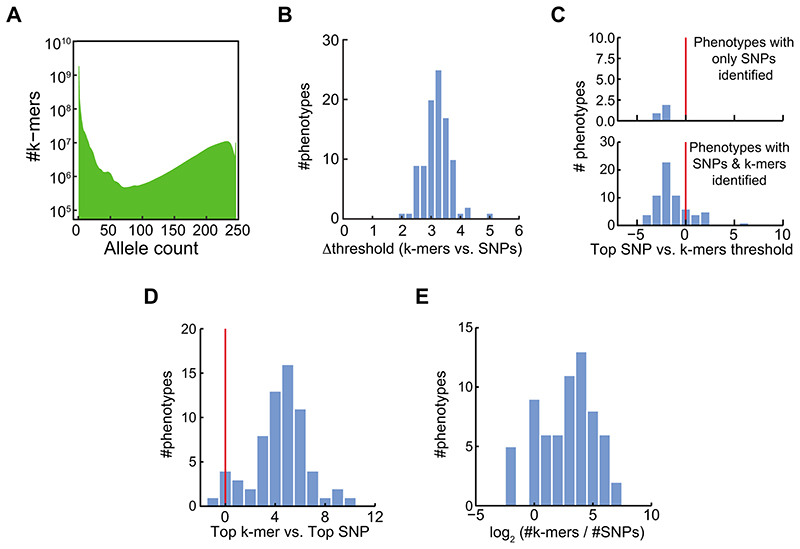
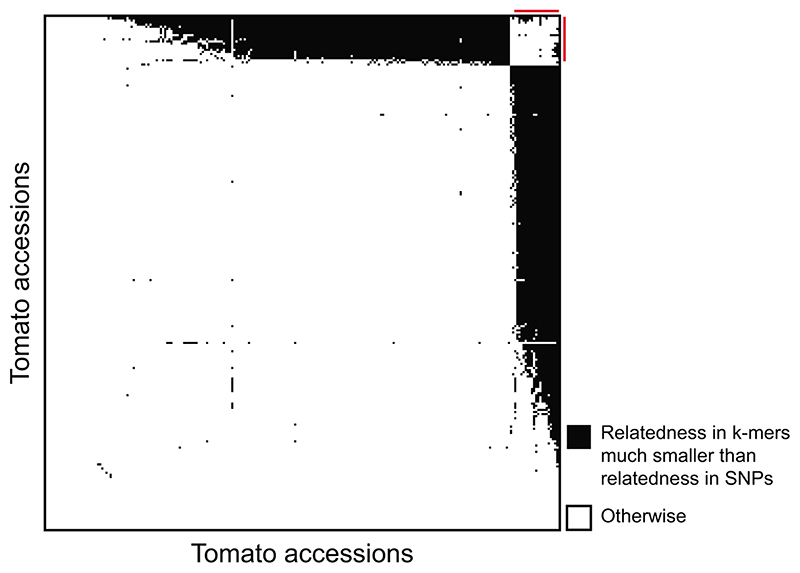
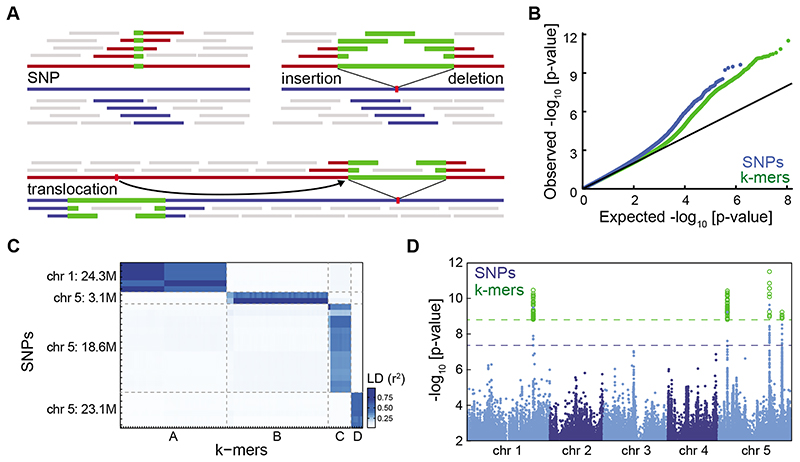
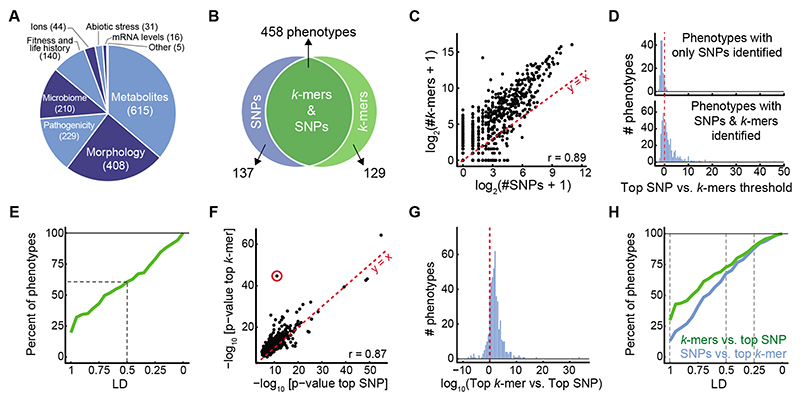
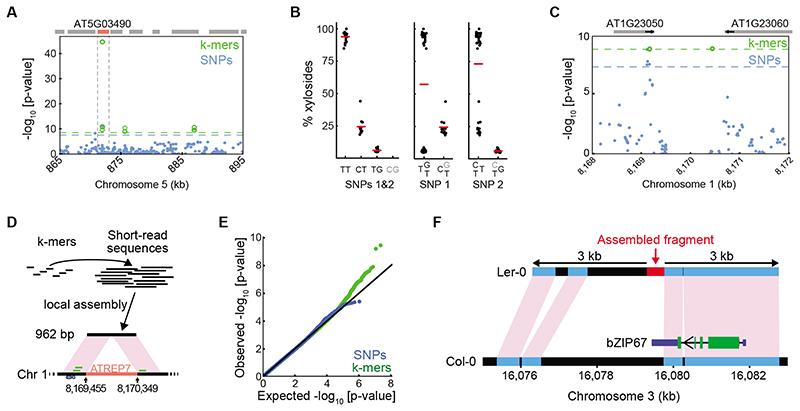
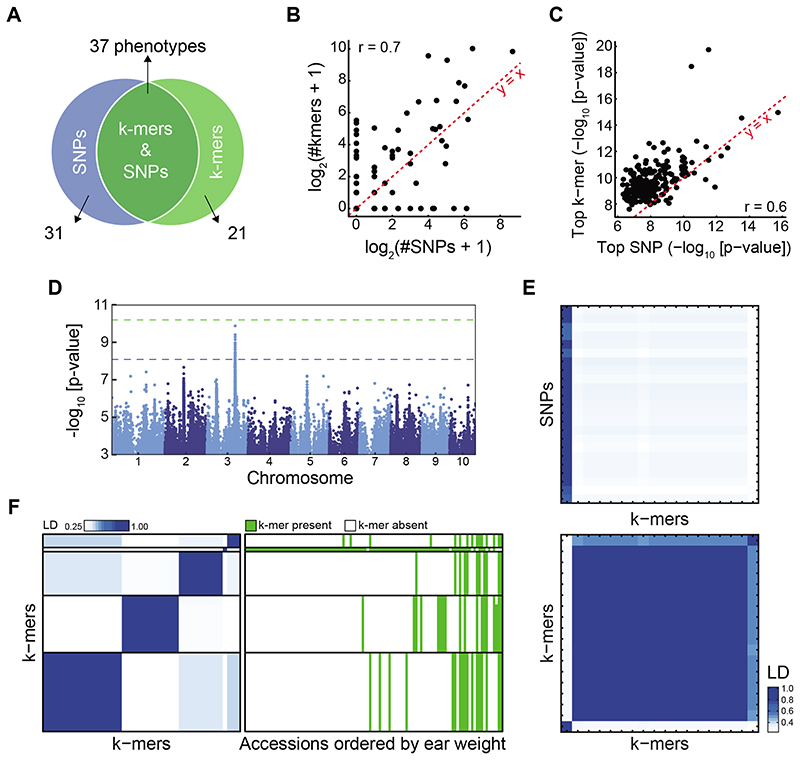
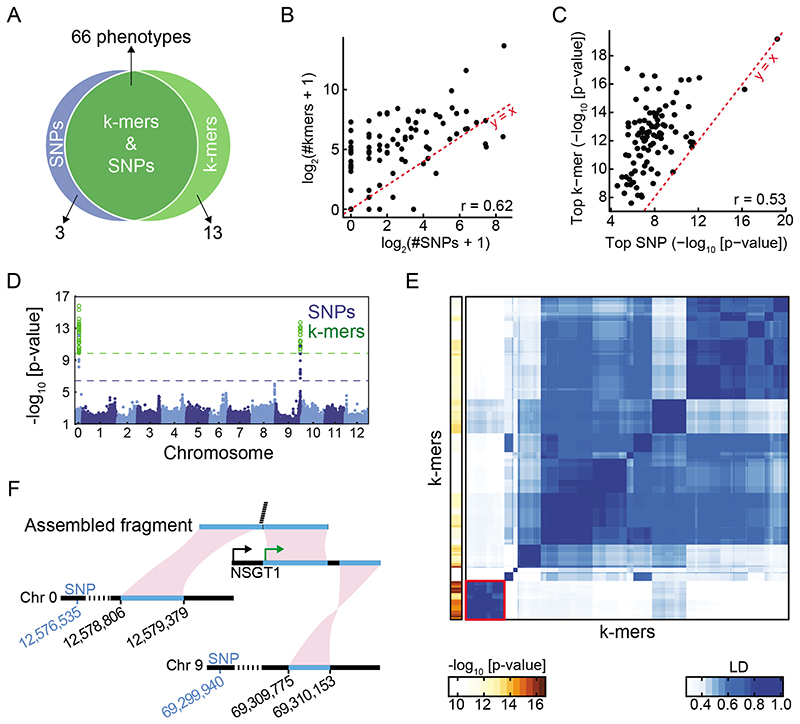
Structural variants and presence/absence polymorphisms are common in plant genomes, yet they are routinely overlooked in genome-wide association studies (GWAS). Here, we expand the type of genetic variants detected in GWAS to include major deletions, insertions and rearrangements. We first use raw sequencing data directly to derive short sequences, k-mers, that mark a broad range of polymorphisms independently of a reference genome. We then link k-mers associated with phenotypes to specific genomic regions. Using this approach, we reanalyzed 2,000 traits in Arabidopsis thaliana, tomato and maize populations. Associations identified with k-mers recapitulate those found with SNPs, but with stronger statistical support. Importantly, we discovered new associations with structural variants and with regions missing from reference genomes. Our results demonstrate the power of performing GWAS before linking sequence reads to specific genomic regions, which allows the detection of a wider range of genetic variants responsible for phenotypic variation.
结构变异和存在/缺失多态性在植物基因组中很常见,但在全基因组关联研究(GWAS)中通常被忽视。在这里,我们将 GWAS 中检测到的遗传变异类型扩展到包括大片段缺失、插入和重排。我们首先使用原始测序数据直接衍生出短序列,即 k-mer,这些 k-mer 可以独立于参考基因组标记广泛的多态性。然后,我们将与表型相关的 k-mer 与特定的基因组区域联系起来。使用这种方法,我们重新分析了拟南芥、番茄和玉米群体中的 2000 个性状。与 k-mer 关联的关联再现了与 SNPs 相关的关联,但具有更强的统计支持。重要的是,我们发现了与结构变异和参考基因组缺失区域相关的新关联。我们的结果表明,在将序列读取与特定基因组区域关联之前进行 GWAS 的强大功能,这允许检测负责表型变异的更广泛的遗传变异。