Perrin B Scott, Tian Ye, Fu Riqiang, Grant Christopher V, Chekmenev Eduard Y, Wieczorek William E, Dao Alexander E, Hayden Robert M, Burzynski Caitlin M, Venable Richard M, Sharma Mukesh, Opella Stanley J, Pastor Richard W, Cotten Myriam L
Laboratory of Computational Biology, National Heart, Lung, and Blood Institute, National Institutes of Health , Bethesda, Maryland 20892, United States.
J Am Chem Soc. 2014 Mar 5;136(9):3491-504. doi: 10.1021/ja411119m. Epub 2014 Jan 22.
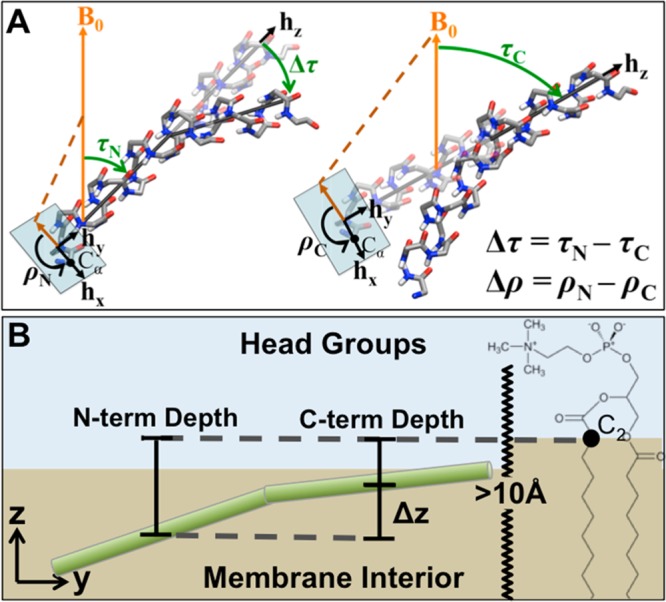
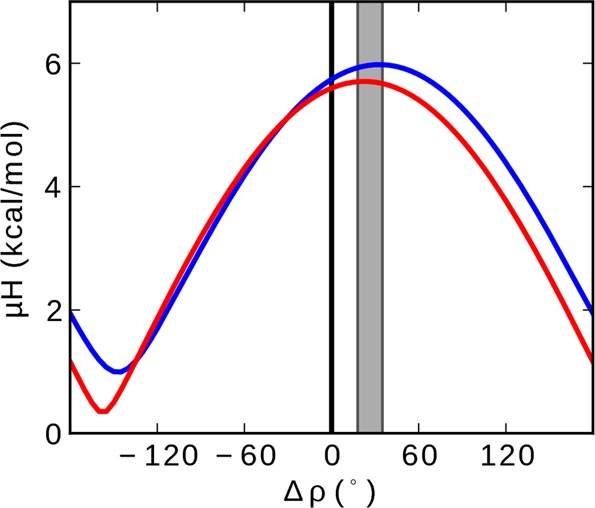
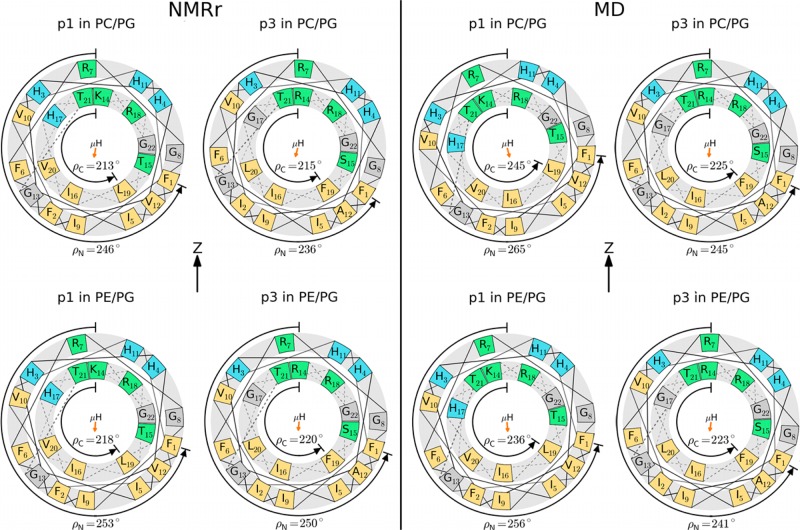
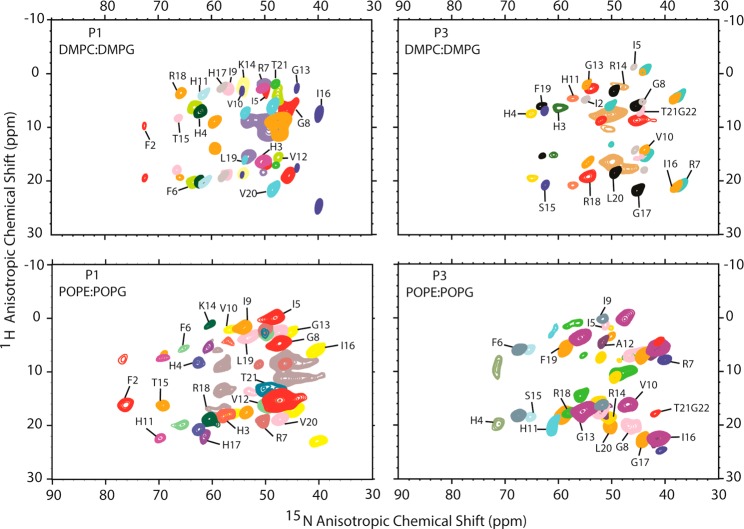
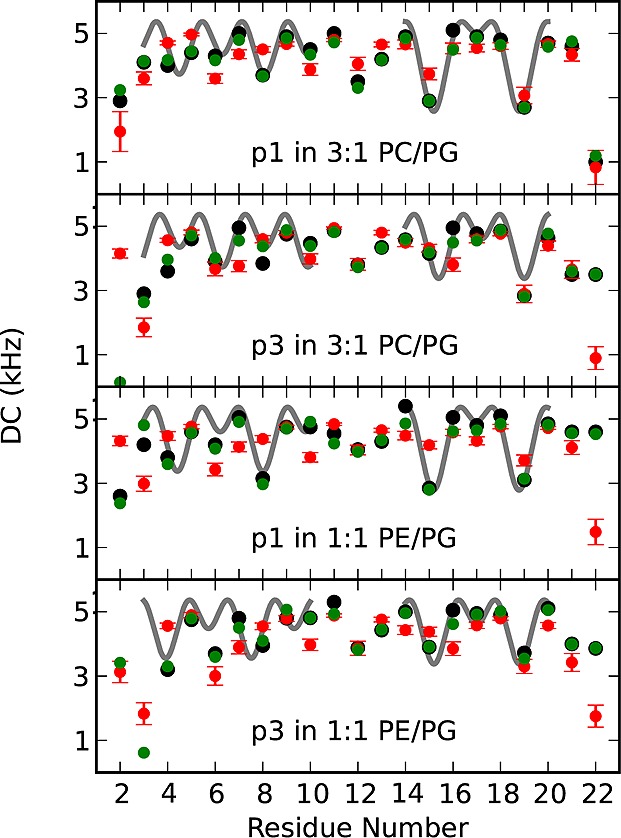
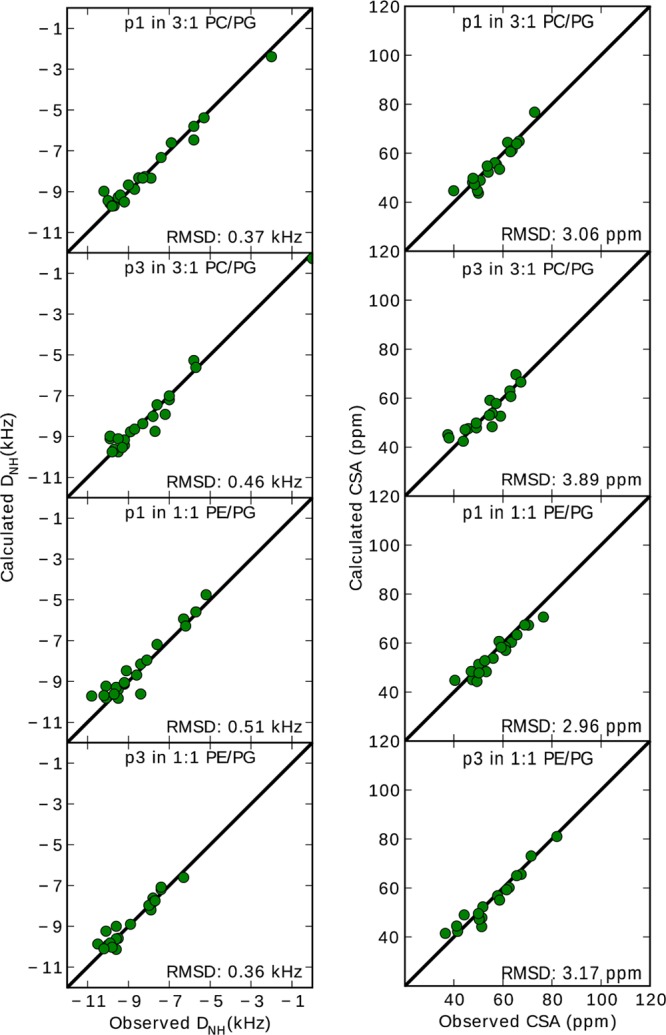
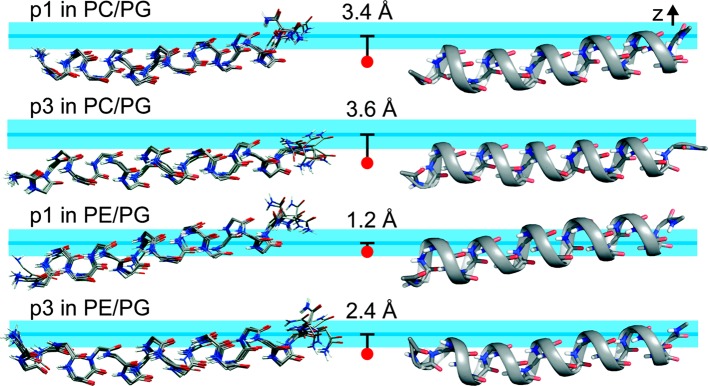
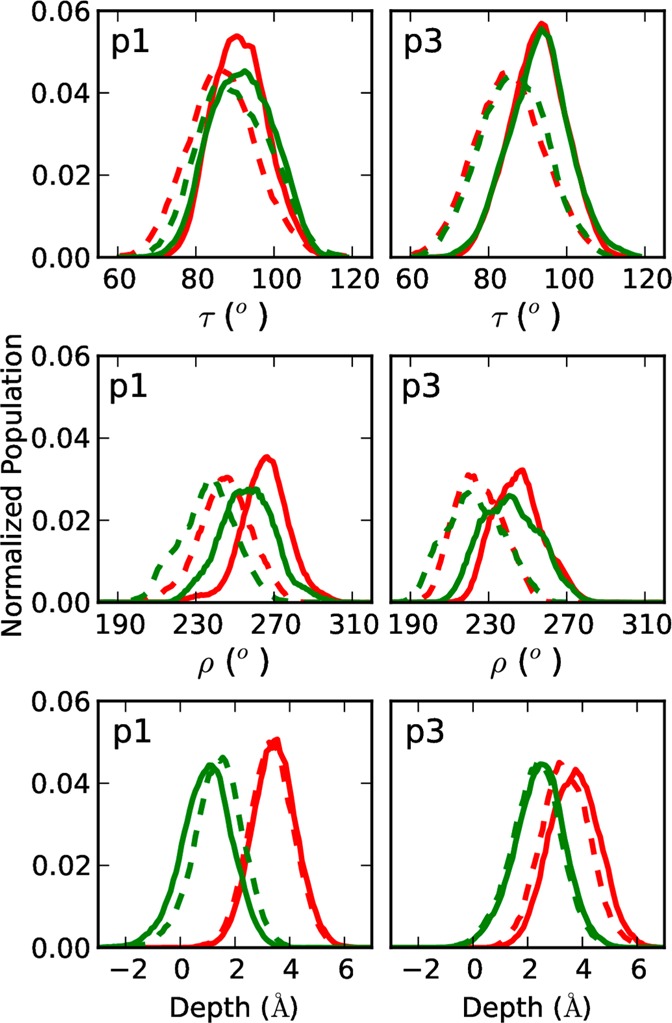
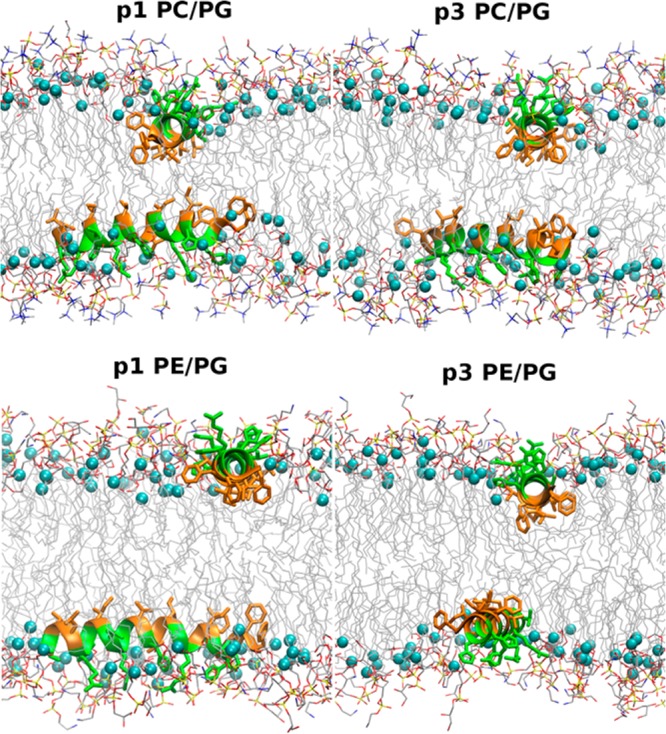
While antimicrobial peptides (AMPs) have been widely investigated as potential therapeutics, high-resolution structures obtained under biologically relevant conditions are lacking. Here, the high-resolution structures of the homologous 22-residue long AMPs piscidin 1 (p1) and piscidin 3 (p3) are determined in fluid-phase 3:1 phosphatidylcholine/phosphatidylglycerol (PC/PG) and 1:1 phosphatidylethanolamine/phosphatidylglycerol (PE/PG) bilayers to identify molecular features important for membrane destabilization in bacterial cell membrane mimics. Structural refinement of (1)H-(15)N dipolar couplings and (15)N chemical shifts measured by oriented sample solid-state NMR and all-atom molecular dynamics (MD) simulations provide structural and orientational information of high precision and accuracy about these interfacially bound α-helical peptides. The tilt of the helical axis, τ, is between 83° and 93° with respect to the bilayer normal for all systems and analysis methods. The average azimuthal rotation, ρ, is 235°, which results in burial of hydrophobic residues in the bilayer. The refined NMR and MD structures reveal a slight kink at G13 that delineates two helical segments characterized by a small difference in their τ angles (<10°) and significant difference in their ρ angles (~25°). Remarkably, the kink, at the end of a G(X)4G motif highly conserved among members of the piscidin family, allows p1 and p3 to adopt ρ angles that maximize their hydrophobic moments. Two structural features differentiate the more potent p1 from p3: p1 has a larger ρ angle and less N-terminal fraying. The peptides have comparable depths of insertion in PC/PG, but p3 is 1.2 Å more deeply inserted than p1 in PE/PG. In contrast to the ideal α-helical structures typically assumed in mechanistic models of AMPs, p1 and p3 adopt disrupted α-helical backbones that correct for differences in the amphipathicity of their N- and C-ends, and their centers of mass lie ~1.2-3.6 Å below the plane defined by the C2 atoms of the lipid acyl chains.
尽管抗菌肽(AMPs)作为潜在治疗药物已得到广泛研究,但在生物学相关条件下获得的高分辨率结构却很匮乏。在此,我们测定了同源的22个残基长的抗菌肽嗜鱼菌素1(p1)和嗜鱼菌素3(p3)在3:1磷脂酰胆碱/磷脂酰甘油(PC/PG)和1:1磷脂酰乙醇胺/磷脂酰甘油(PE/PG)双分子层液相中的高分辨率结构,以确定在细菌细胞膜模拟物中对膜去稳定化重要的分子特征。通过定向样品固态核磁共振(NMR)测量的(1)H - (15)N偶极耦合和(15)N化学位移以及全原子分子动力学(MD)模拟的结构优化,提供了关于这些界面结合的α - 螺旋肽的高精度和准确性的结构及取向信息。对于所有系统和分析方法,螺旋轴相对于双分子层法线的倾斜角τ在83°至93°之间。平均方位角旋转ρ为235°,这导致疏水残基埋入双分子层中。优化后的NMR和MD结构显示在G13处有一个轻微的扭结,它划分出两个螺旋段,其特征在于它们的τ角差异较小(<10°),而ρ角差异较大(约25°)。值得注意的是,在嗜鱼菌素家族成员中高度保守的G(X)4G基序末端的扭结,使p1和p3能够采用使它们的疏水矩最大化的ρ角。有两个结构特征区分了活性更强的p1和p3:p1具有更大的ρ角且N端磨损更少。这些肽在PC/PG中的插入深度相当,但在PE/PG中p3比p1深插入1.2 Å。与AMPs机制模型中通常假设的理想α - 螺旋结构相反,p1和p3采用了破坏的α - 螺旋主链,以校正其N端和C端两亲性的差异,并且它们的质心位于脂质酰链C2原子定义的平面下方约1.2 - 3.6 Å处。