Agriculture and AgriFood Canada, 107 Science Place, S7N 0X2, Saskatoon, SK, Canada.
Microbiome. 2013 Aug 15;1(1):23. doi: 10.1186/2049-2618-1-23.
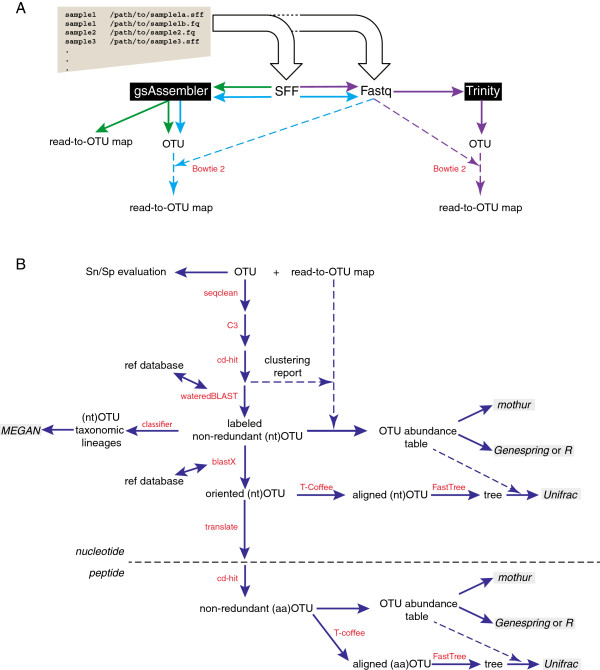
Formation of operational taxonomic units (OTU) is a common approach to data aggregation in microbial ecology studies based on amplification and sequencing of individual gene targets. The de novo assembly of OTU sequences has been recently demonstrated as an alternative to widely used clustering methods, providing robust information from experimental data alone, without any reliance on an external reference database.
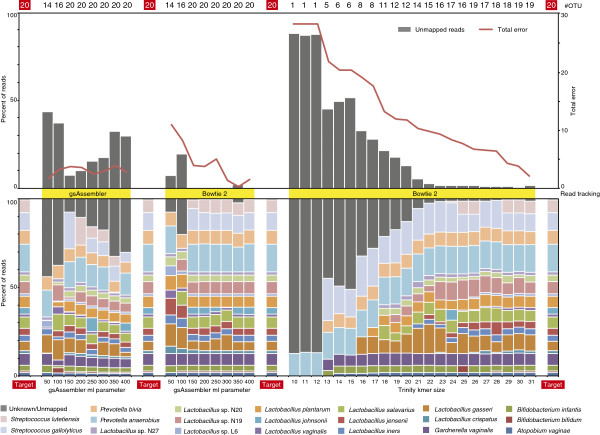
Here we introduce mPUMA (microbial Profiling Using Metagenomic Assembly, http://mpuma.sourceforge.net), a software package for identification and analysis of protein-coding barcode sequence data. It was developed originally for Cpn60 universal target sequences (also known as GroEL or Hsp60). Using an unattended process that is independent of external reference sequences, mPUMA forms OTUs by DNA sequence assembly and is capable of tracking OTU abundance. mPUMA processes microbial profiles both in terms of the direct DNA sequence as well as in the translated amino acid sequence for protein coding barcodes. By forming OTUs and calculating abundance through an assembly approach, mPUMA is capable of generating inputs for several popular microbiota analysis tools. Using SFF data from sequencing of a synthetic community of Cpn60 sequences derived from the human vaginal microbiome, we demonstrate that mPUMA can faithfully reconstruct all expected OTU sequences and produce compositional profiles consistent with actual community structure.
mPUMA enables analysis of microbial communities while empowering the discovery of novel organisms through OTU assembly.
基于个体基因靶标的扩增和测序的操作分类单元(OTU)的形成是微生物生态学研究中数据聚合的常用方法。OTU 序列的从头组装最近已被证明是广泛使用的聚类方法的替代方法,仅从实验数据中提供稳健的信息,而无需依赖外部参考数据库。
在这里,我们介绍了 mPUMA(使用宏基因组组装进行微生物分析,http://mpuma.sourceforge.net),这是一个用于鉴定和分析蛋白质编码条码序列数据的软件包。它最初是为 Cpn60 通用目标序列(也称为 GroEL 或 Hsp60)开发的。mPUMA 通过 DNA 序列组装形成 OTU,并且能够跟踪 OTU 的丰度,使用无需外部参考序列的无人值守过程。mPUMA 以直接 DNA 序列和蛋白质编码条码的翻译氨基酸序列两种方式处理微生物分布。通过形成 OTU 并通过组装方法计算丰度,mPUMA 能够为几种流行的微生物组分析工具生成输入。使用源自人类阴道微生物组的 Cpn60 序列的合成群落测序的 SFF 数据,我们证明 mPUMA 能够忠实地重建所有预期的 OTU 序列,并产生与实际群落结构一致的组成分布。
mPUMA 能够在通过 OTU 组装发现新的生物体的同时分析微生物群落。