Garrett Scott H, Somji Seema, Sens Donald A, Zhang Ke K
Department of Pathology, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, North Dakota, United States of America.
PLoS One. 2014 Jan 22;9(1):e85614. doi: 10.1371/journal.pone.0085614. eCollection 2014.
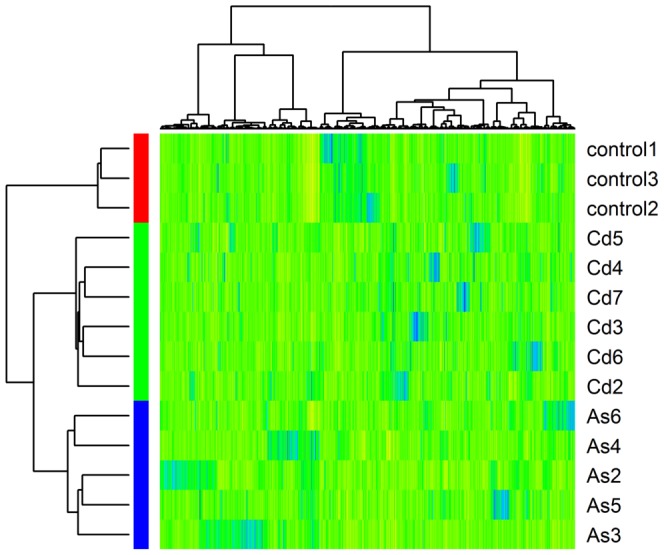
Many toxic environmental agents such as cadmium and arsenic are found to be epidemiologically linked to increasing rates of cancers. In vitro studies show that these toxic agents induced malignant transformation in human cells. It is not clear whether such malignant transformation induced by a single toxic agent is driven by a common set of genes. Although the advancement of high-throughput technology has facilitated the profiling of global gene expression, it remains a question whether the sample size is sufficient to identify this common driver gene set.
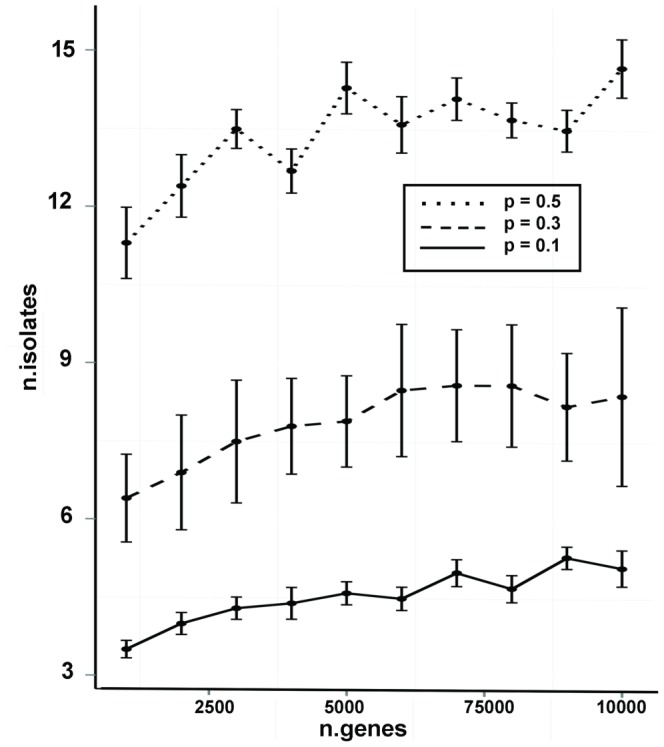
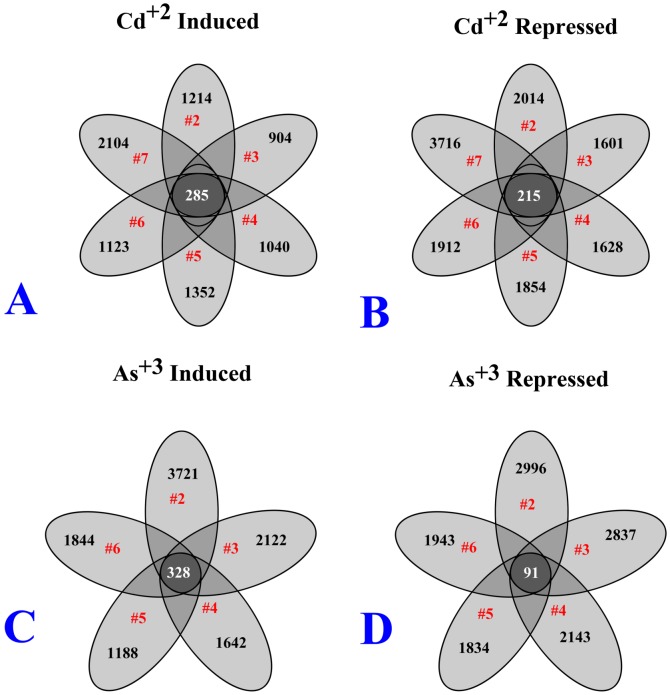
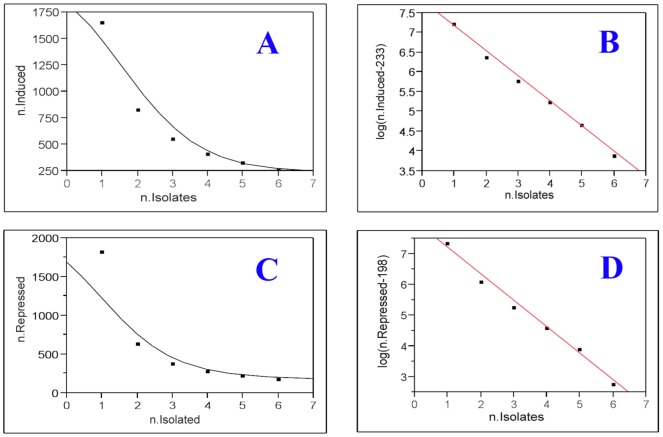
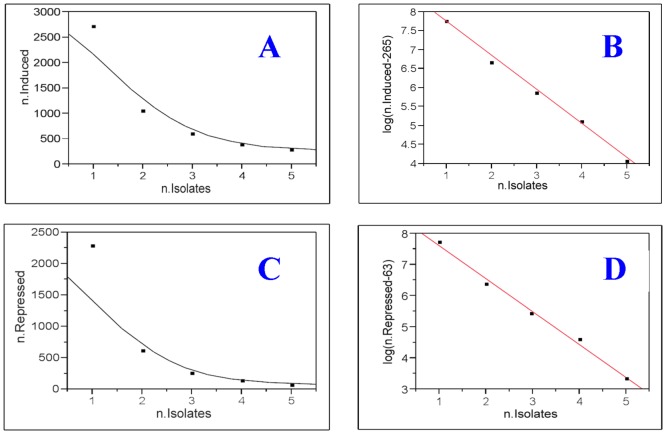
We have developed a statistical method, SOFLR, to predict the number of common activated genes using a limited number of microarray samples. We conducted two case studies, cadmium and arsenic transformed human urothelial cells. Our method is able to precisely predict the number of stably induced and repressed genes and the number of samples to identify the common activated genes. The number of independent transformed isolates required for identifying the common activated genes is also estimated. The simulation study further validated our method and identified the important parameters in this analysis.
Our method predicts the degree of similarity and diversity during cell malignant transformation by a single toxic agent. The results of our case studies imply the existence of common driver and passenger gene sets in toxin-induced transformation. Using a pilot study with small sample size, this method also helps microarray experimental design by determining the number of samples required to identify the common activated gene set.
许多有毒环境因子,如镉和砷,在流行病学上被发现与癌症发病率的上升有关。体外研究表明,这些有毒因子可诱导人类细胞发生恶性转化。目前尚不清楚单一有毒因子诱导的这种恶性转化是否由一组共同的基因驱动。尽管高通量技术的进步促进了全球基因表达谱的分析,但样本量是否足以识别这一共同的驱动基因集仍是一个问题。
我们开发了一种统计方法SOFLR,用于使用有限数量的微阵列样本预测共同激活基因的数量。我们进行了两个案例研究,即镉和砷转化的人类尿道上皮细胞。我们的方法能够精确预测稳定诱导和抑制基因的数量以及识别共同激活基因所需的样本数量。还估计了识别共同激活基因所需的独立转化分离株的数量。模拟研究进一步验证了我们的方法,并确定了该分析中的重要参数。
我们的方法预测了单一有毒因子诱导细胞恶性转化过程中的相似性和多样性程度。我们案例研究的结果表明,在毒素诱导的转化中存在共同的驱动基因集和乘客基因集。通过使用小样本量的初步研究,该方法还通过确定识别共同激活基因集所需的样本数量,帮助进行微阵列实验设计。