Department of Pathology, School of Medicine and Health Sciences, University of North Dakota, Grand Forks, ND 58201, USA.
BMC Genomics. 2011 Dec 23;12 Suppl 5(Suppl 5):S10. doi: 10.1186/1471-2164-12-S5-S10.
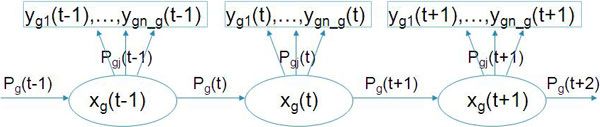
The recent advancement in array CGH (aCGH) research has significantly improved tumor identification using DNA copy number data. A number of unsupervised learning methods have been proposed for clustering aCGH samples. Two of the major challenges for developing aCGH sample clustering are the high spatial correlation between aCGH markers and the low computing efficiency. A mixture hidden Markov model based algorithm was developed to address these two challenges.
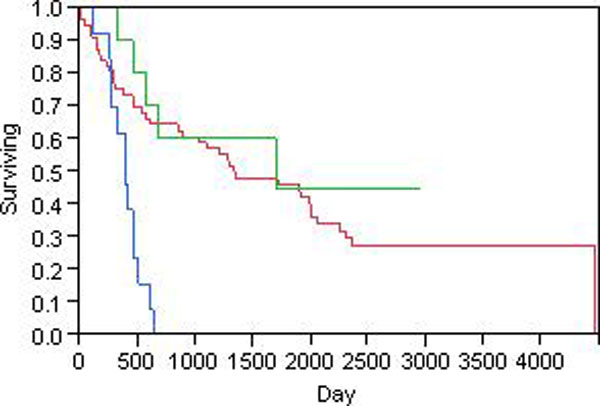
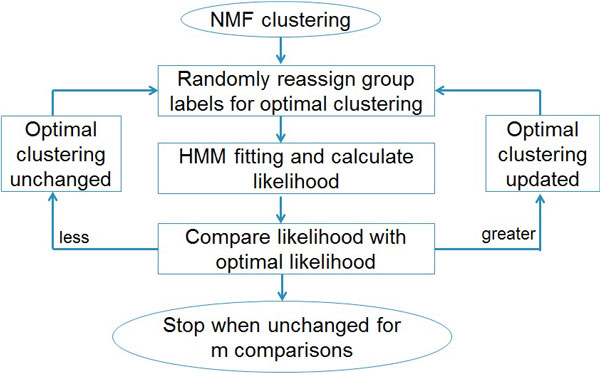
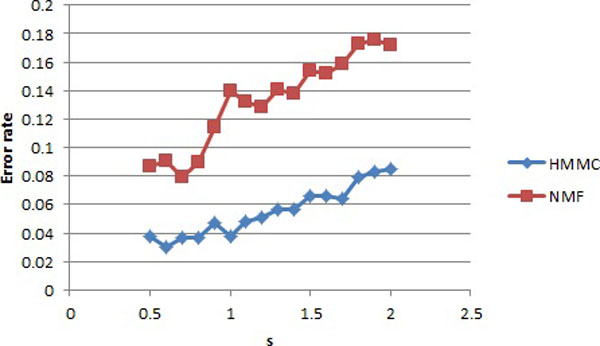
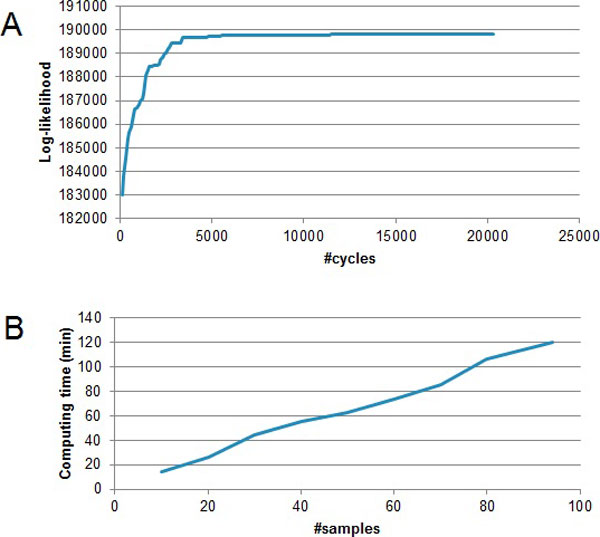
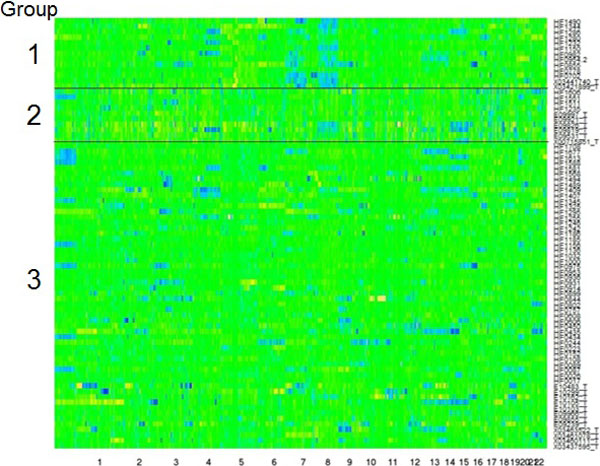
The hidden Markov model (HMM) was used to model the spatial correlation between aCGH markers. A fast clustering algorithm was implemented and real data analysis on glioma aCGH data has shown that it converges to the optimal cluster rapidly and the computation time is proportional to the sample size. Simulation results showed that this HMM based clustering (HMMC) method has a substantially lower error rate than NMF clustering. The HMMC results for glioma data were significantly associated with clinical outcomes.
We have developed a fast clustering algorithm to identify tumor subtypes based on DNA copy number aberrations. The performance of the proposed HMMC method has been evaluated using both simulated and real aCGH data. The software for HMMC in both R and C++ is available in ND INBRE website http://ndinbre.org/programs/bioinformatics.php.
近年来 array CGH(aCGH)研究的进展显著提高了使用 DNA 拷贝数数据进行肿瘤识别的能力。已经提出了许多无监督学习方法来对 aCGH 样本进行聚类。开发 aCGH 样本聚类的两个主要挑战是 aCGH 标记之间的高度空间相关性和低计算效率。基于混合隐马尔可夫模型的算法被开发出来以解决这两个挑战。
隐马尔可夫模型(HMM)用于对 aCGH 标记之间的空间相关性进行建模。实现了一种快速聚类算法,对 glioma aCGH 数据的实际数据分析表明,它能够快速收敛到最优聚类,并且计算时间与样本大小成正比。模拟结果表明,这种基于 HMM 的聚类(HMMC)方法的错误率明显低于 NMF 聚类。glioma 数据的 HMMC 结果与临床结果显著相关。
我们开发了一种快速聚类算法,基于 DNA 拷贝数异常来识别肿瘤亚型。使用模拟和真实的 aCGH 数据评估了所提出的 HMMC 方法的性能。R 和 C++中的 HMMC 软件可在 ND INBRE 网站 http://ndinbre.org/programs/bioinformatics.php 上获得。