Genome Research Center, NODAI Research Institute, Tokyo University of Agriculture, Setagaya, Tokyo, Japan.
Genetic Resources Center, National Institute of Agrobiological Sciences, Tsukuba, Ibaraki, Japan.
PLoS One. 2014 Jan 21;9(1):e86312. doi: 10.1371/journal.pone.0086312. eCollection 2014.
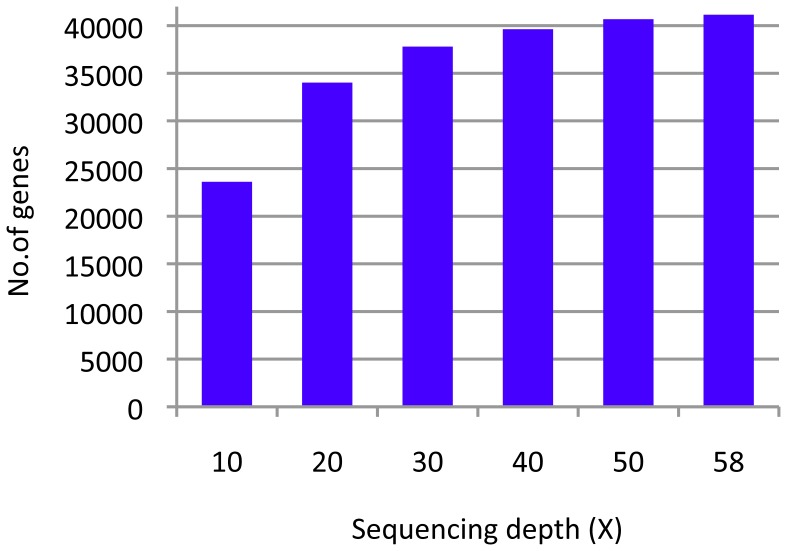
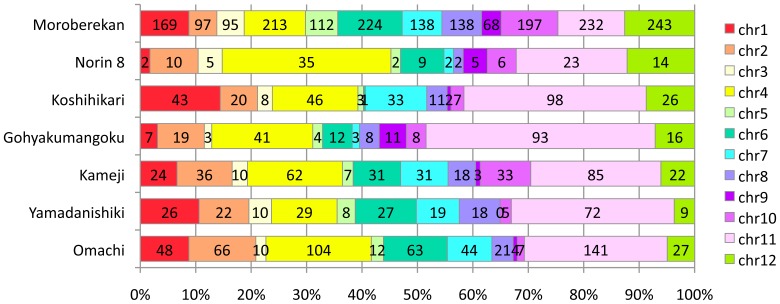
Elucidation of the rice genome is expected to broaden our understanding of genes related to the agronomic characteristics and the genetic relationship among cultivars. In this study, we conducted whole-genome sequencings of 6 cultivars, including 5 temperate japonica cultivars and 1 tropical japonica cultivar (Moroberekan), by using next-generation sequencing (NGS) with Nipponbare genome as a reference. The temperate japonica cultivars contained 2 sake brewing (Yamadanishiki and Gohyakumangoku), 1 landrace (Kameji), and 2 modern cultivars (Koshihikari and Norin 8). Almost >83% of the whole genome sequences of the Nipponbare genome could be covered by sequenced short-reads of each cultivar, including Omachi, which has previously been reported to be a temperate japonica cultivar. Numerous single nucleotide polymorphisms (SNPs), insertions, and deletions were detected among the various cultivars and the Nipponbare genomes. Comparison of SNPs detected in each cultivar suggested that Moroberekan had 5-fold more SNPs than the temperate japonica cultivars. Success of the 2 approaches to improve the efficacy of sequence data by using NGS revealed that sequencing depth was directly related to sequencing coverage of coding DNA sequences: in excess of 30× genome sequencing was required to cover approximately 80% of the genes in the rice genome. Further, the contigs prepared using the assembly of unmapped reads could increase the value of NGS short-reads and, consequently, cover previously unavailable sequences. These approaches facilitated the identification of new genes in coding DNA sequences and the increase of mapping efficiency in different regions. The DNA polymorphism information between the 7 cultivars and Nipponbare are available at NGRC_Rices_Build1.0 (http://www.nodai-genome.org/oryza_sativa_en.html).
阐明水稻基因组有望加深我们对与农艺性状相关的基因以及品种间遗传关系的理解。在这项研究中,我们利用下一代测序(NGS)技术,以日本晴基因组作为参考,对 6 个品种(包括 5 个温带粳稻品种和 1 个热带粳稻品种 Moroberekan)进行了全基因组测序。这 5 个温带粳稻品种包括 2 个清酒酿造品种(Yamadanishiki 和 Gohyakumangoku)、1 个地方品种(Kameji)和 2 个现代品种(Koshihikari 和 Norin 8)。每个品种的测序短读序列几乎都能覆盖日本晴基因组的 83%以上,包括此前被报道为温带粳稻品种的 Omachi。在不同品种和日本晴基因组之间检测到了大量的单核苷酸多态性(SNP)、插入和缺失。在每个品种中检测到的 SNP 的比较表明,Moroberekan 的 SNP 数量是温带粳稻品种的 5 倍。通过使用 NGS 提高序列数据效率的两种方法的成功表明,测序深度与编码 DNA 序列的测序覆盖度直接相关:需要超过 30×的基因组测序才能覆盖水稻基因组中约 80%的基因。此外,使用未映射读组装制备的 contigs 可以提高 NGS 短读的价值,并覆盖以前无法获得的序列。这些方法有助于在编码 DNA 序列中鉴定新基因,并提高不同区域的映射效率。这 7 个品种与日本晴之间的 DNA 多态性信息可在 NGRC_Rices_Build1.0 (http://www.nodai-genome.org/oryza_sativa_en.html)上获取。