Reimer Lorenz C, Spura Jana, Schmidt-Hohagen Kerstin, Schomburg Dietmar
Department of Bioinformatics and Biochemistry, Technische Universität Braunschweig, Braunschweig, Germany.
PLoS One. 2014 Feb 4;9(2):e86799. doi: 10.1371/journal.pone.0086799. eCollection 2014.
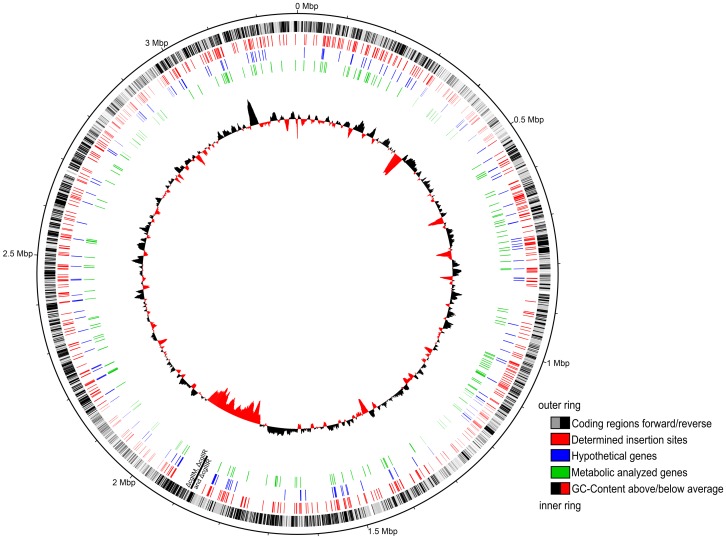
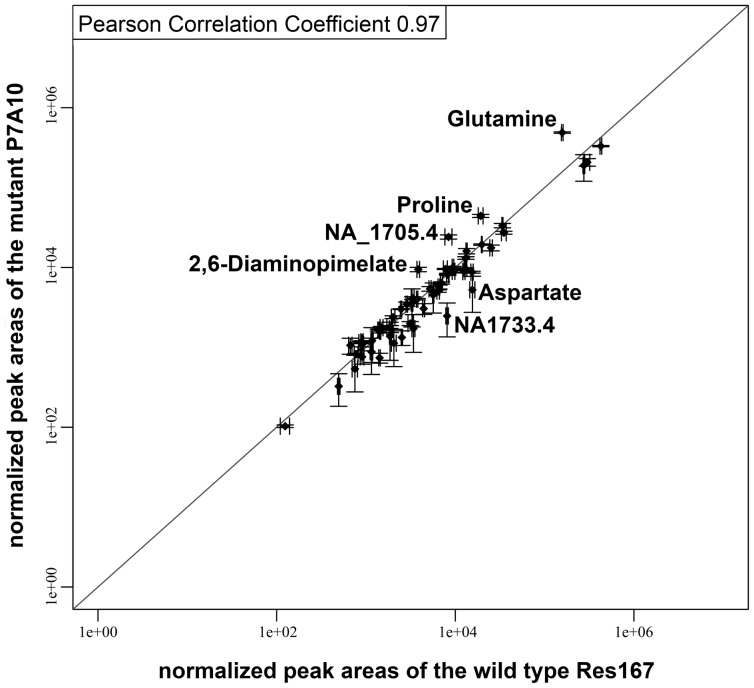
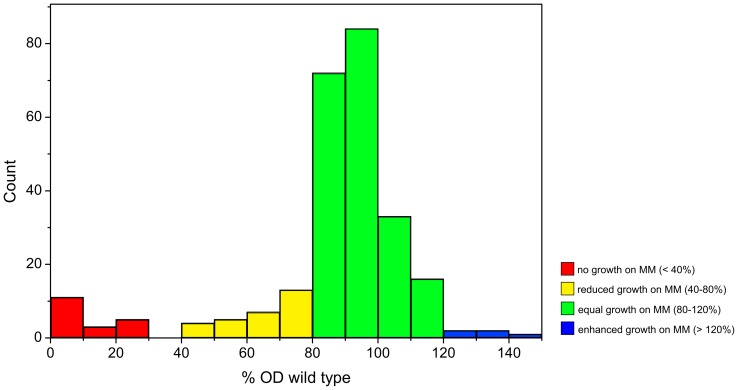
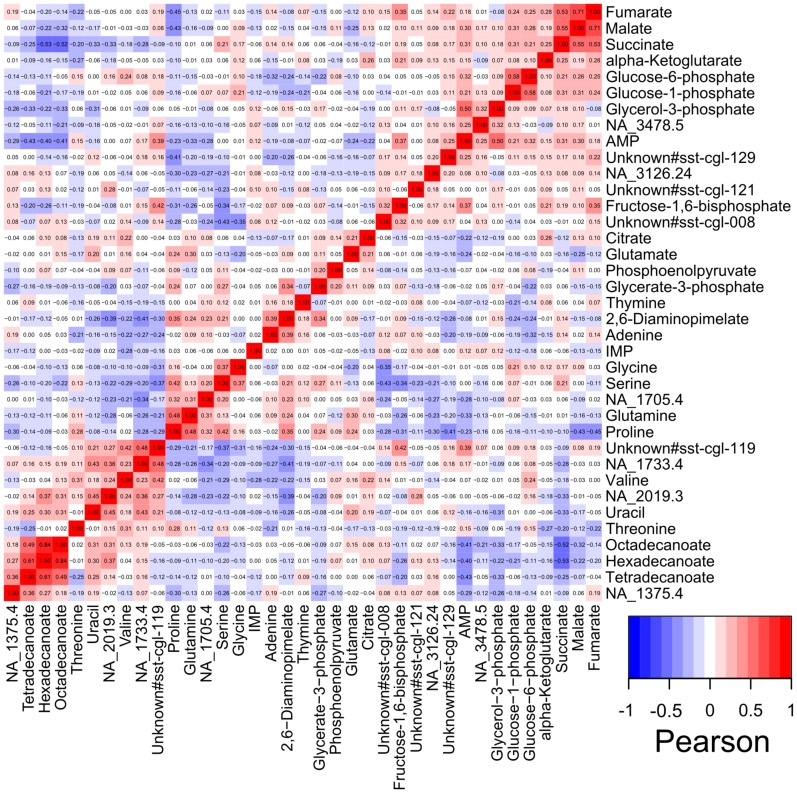
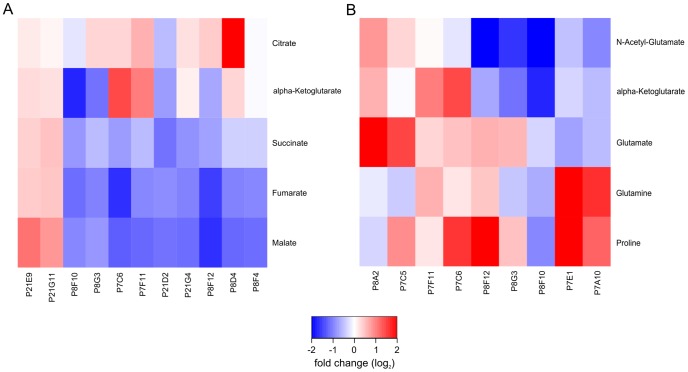
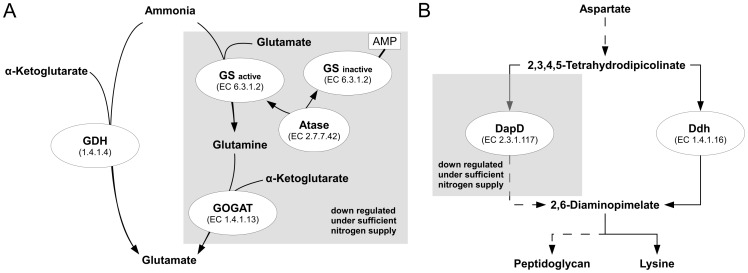
Due to impressive achievements in genomic research, the number of genome sequences has risen quickly, followed by an increasing number of genes with unknown or hypothetical function. This strongly calls for development of high-throughput methods in the fields of transcriptomics, proteomics and metabolomics. Of these platforms, metabolic profiling has the strongest correlation with the phenotype. We previously published a high-throughput metabolic profiling method for C. glutamicum as well as the automatic GC/MS processing software MetaboliteDetector. Here, we added a high-throughput transposon insertion determination for our C. glutamicum mutant library. The combination of these methods allows the parallel analysis of genotype/phenotype correlations for a large number of mutants. In a pilot project we analyzed the insertion points of 722 transposon mutants and found that 36% of the affected genes have unknown functions. This underlines the need for further information gathered by high-throughput techniques. We therefore measured the metabolic profiles of 258 randomly chosen mutants. The MetaboliteDetector software processed this large amount of GC/MS data within a few hours with a low relative error of 11.5% for technical replicates. Pairwise correlation analysis of metabolites over all genotypes showed dependencies of known and unknown metabolites. For a first insight into this large data set, a screening for interesting mutants was done by a pattern search, focusing on mutants with changes in specific pathways. We show that our transposon mutant library is not biased with respect to insertion points. A comparison of the results for specific mutants with previously published metabolic results on a deletion mutant of the same gene confirmed the concept of high-throughput metabolic profiling. Altogether the described method could be applied to whole mutant libraries and thereby help to gain comprehensive information about genes with unknown, hypothetical and known functions.
由于基因组研究取得了令人瞩目的成就,基因组序列的数量迅速增加,随之而来的是功能未知或假设的基因数量也在不断增加。这强烈呼吁在转录组学、蛋白质组学和代谢组学领域开发高通量方法。在这些平台中,代谢谱分析与表型的相关性最强。我们之前发表了一种针对谷氨酸棒杆菌的高通量代谢谱分析方法以及自动气相色谱/质谱处理软件MetaboliteDetector。在此,我们为我们的谷氨酸棒杆菌突变体文库添加了高通量转座子插入测定。这些方法的结合允许对大量突变体的基因型/表型相关性进行并行分析。在一个试点项目中,我们分析了722个转座子突变体的插入点,发现36%的受影响基因功能未知。这突出了通过高通量技术收集更多信息的必要性。因此,我们测量了258个随机选择的突变体的代谢谱。MetaboliteDetector软件在几个小时内处理了大量的气相色谱/质谱数据,技术重复的相对误差低至11.5%。对所有基因型的代谢物进行成对相关性分析,显示了已知和未知代谢物之间的依赖性。为了初步了解这个庞大的数据集,通过模式搜索对有趣的突变体进行了筛选,重点关注特定途径发生变化的突变体。我们表明,我们的转座子突变体文库在插入点方面没有偏差。将特定突变体的结果与之前发表的同一基因缺失突变体的代谢结果进行比较,证实了高通量代谢谱分析的概念。总之,所描述的方法可以应用于整个突变体文库,从而有助于获得关于功能未知、假设和已知基因的全面信息。