Division of Biostatistics, Washington University School of Medicine in St. Louis St. Louis, MO, USA.
Front Genet. 2014 Jan 30;5:9. doi: 10.3389/fgene.2014.00009. eCollection 2014.
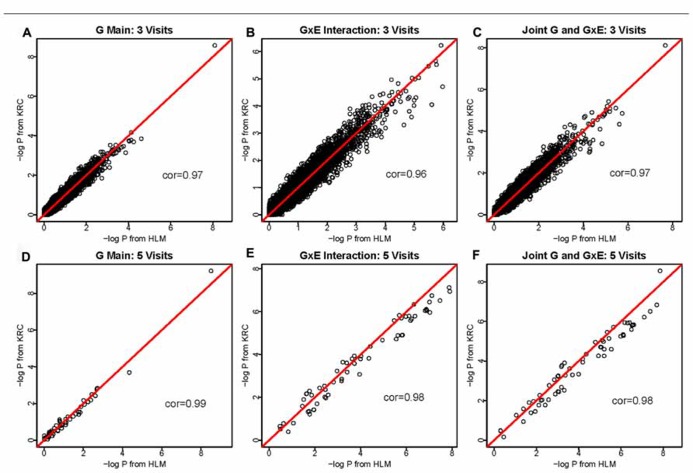
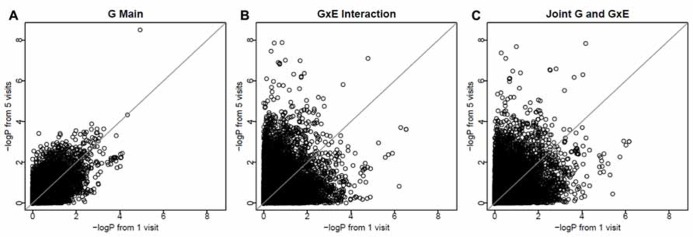
Gene-environment interaction (GEI) analysis can potentially enhance gene discovery for common complex traits. However, genome-wide interaction analysis is computationally intensive. Moreover, analysis of longitudinal data in families is much more challenging due to the two sources of correlations arising from longitudinal measurements and family relationships. GWIS of longitudinal family data can be a computational bottleneck. Therefore, we compared two methods for analysis of longitudinal family data: a methodologically sound but computationally demanding method using the Kronecker model (KRC) and a computationally more forgiving method using the hierarchical linear model (HLM). The KRC model uses a Kronecker product of an unstructured matrix for correlations among repeated measures (longitudinal) and a compound symmetry matrix for correlations within families at a given visit. The HLM uses an autoregressive covariance matrix for correlations among repeated measures and a random intercept for familial correlations. We compared the two methods using the longitudinal Framingham heart study (FHS) SHARe data. Specifically, we evaluated SNP-alcohol (amount of alcohol consumption) interaction effects on high density lipoprotein cholesterol (HDLC). Keeping the prohibitive computational burden of KRC in mind, we limited the analysis to chromosome 16, where preliminary cross-sectional analysis yielded some interesting results. Our first important finding was that the HLM provided very comparable results but was remarkably faster than the KRC, making HLM the method of choice. Our second finding was that longitudinal analysis provided smaller P-values, thus leading to more significant results, than cross-sectional analysis. This was particularly pronounced in identifying GEIs. We conclude that longitudinal analysis of GEIs is more powerful and that the HLM method is an optimal method of choice as compared to the computationally (prohibitively) intensive KRC method.
基因-环境交互作用(GEI)分析有可能增强常见复杂性状的基因发现。然而,全基因组交互分析计算量很大。此外,由于纵向测量和家庭关系产生的两种相关性来源,对家庭的纵向数据进行分析更加具有挑战性。对纵向家庭数据进行 GWIS 可能是计算上的瓶颈。因此,我们比较了两种分析纵向家庭数据的方法:一种是使用 Kronecker 模型(KRC)的方法学上合理但计算上要求很高的方法,另一种是使用层次线性模型(HLM)的计算上更宽容的方法。KRC 模型使用重复测量(纵向)之间相关性的非结构化矩阵的 Kronecker 积和给定访问内家庭之间相关性的复合对称矩阵。HLM 使用重复测量之间的自回归协方差矩阵和家族相关性的随机截距。我们使用纵向弗雷明汉心脏研究(FHS)SHARe 数据比较了这两种方法。具体来说,我们评估了 SNP-酒精(饮酒量)相互作用对高密度脂蛋白胆固醇(HDLC)的影响。考虑到 KRC 的计算负担很大,我们将分析仅限于染色体 16,其中初步的横截面分析产生了一些有趣的结果。我们的第一个重要发现是,HLM 提供了非常可比的结果,但比 KRC 快得多,这使得 HLM 成为首选方法。我们的第二个发现是,纵向分析提供了更小的 P 值,从而导致更显著的结果,而不是横截面分析。在识别 GEI 方面尤其明显。我们得出的结论是,纵向分析 GEI 更有效,与计算上(过度)密集的 KRC 方法相比,HLM 方法是一种最佳选择方法。