Neurogenetics Program and Department of Neurology, David Geffen School of Medicine, University of California, Los Angeles, 90095 Los Angeles, CA, USA.
Mol Autism. 2014 Feb 17;5(1):13. doi: 10.1186/2040-2392-5-13.
Autism spectrum disorders (ASDs) are male-biased and genetically heterogeneous. While sequencing of sporadic cases has identified de novo risk variants, the heritable genetic contribution and mechanisms driving the male bias are less understood. Here, we aimed to identify familial and sex-differential risk loci in the largest available, uniformly ascertained, densely genotyped sample of multiplex ASD families from the Autism Genetics Resource Exchange (AGRE), and to compare results with earlier findings from AGRE.
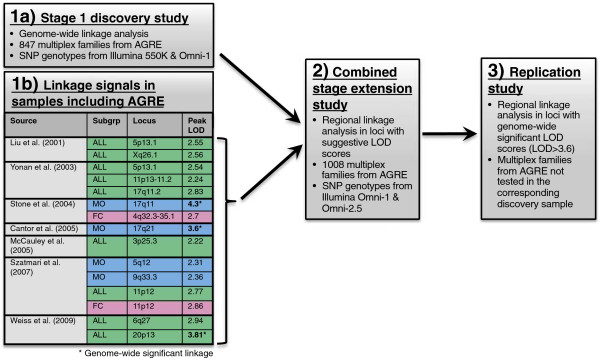
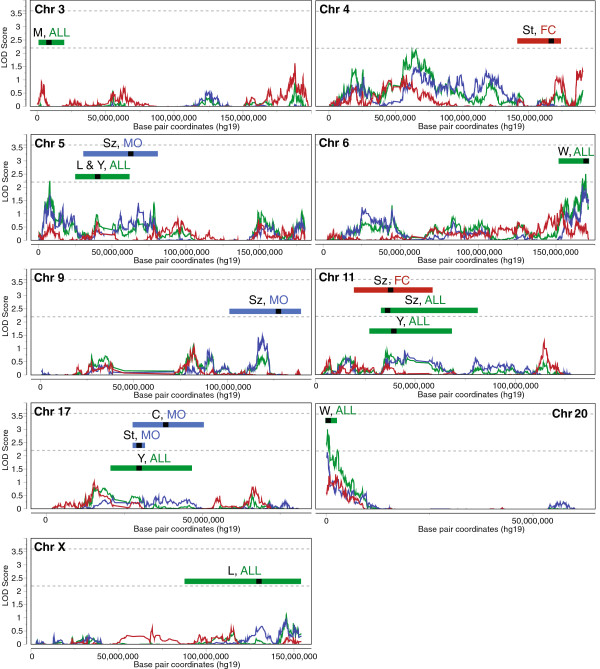
From a total sample of 1,008 multiplex families, we performed genome-wide, non-parametric linkage analysis in a discovery sample of 847 families, and separately on subsets of families with only male, affected children (male-only, MO) or with at least one female, affected child (female-containing, FC). Loci showing evidence for suggestive linkage (logarithm of odds ≥2.2) in this discovery sample, or in previous AGRE samples, were re-evaluated in an extension study utilizing all 1,008 available families. For regions with genome-wide significant linkage signal in the discovery stage, those families not included in the corresponding discovery sample were then evaluated for independent replication of linkage. Association testing of common single nucleotide polymorphisms (SNPs) was also performed within suggestive linkage regions.
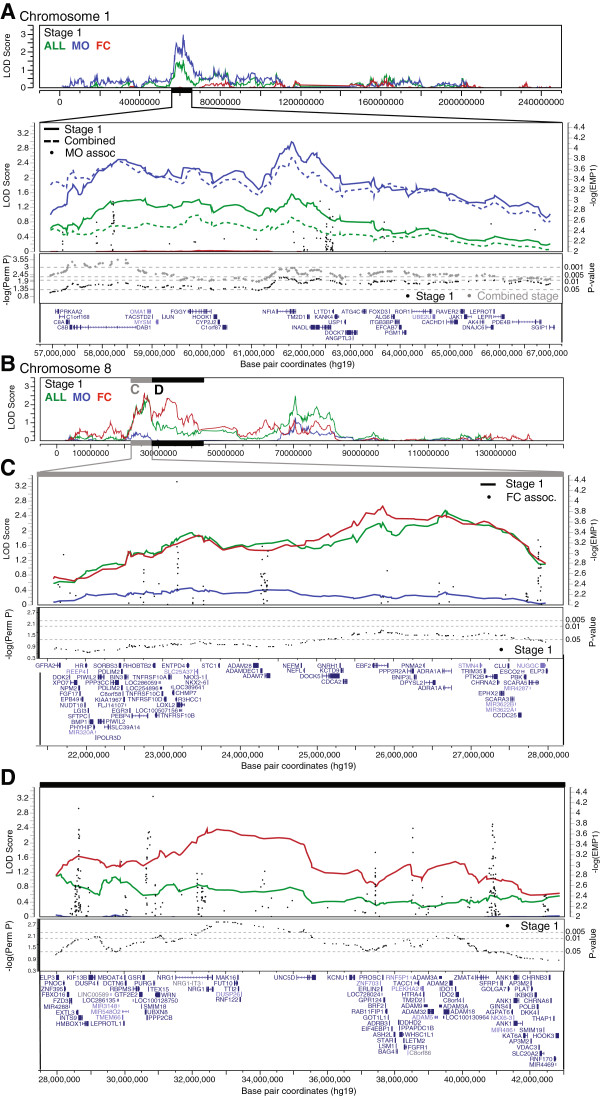
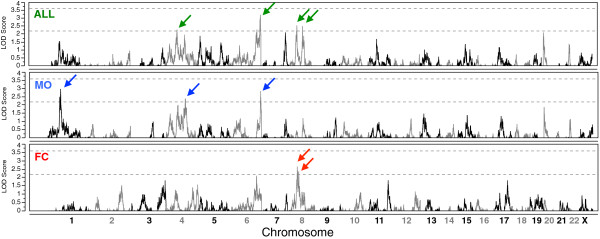
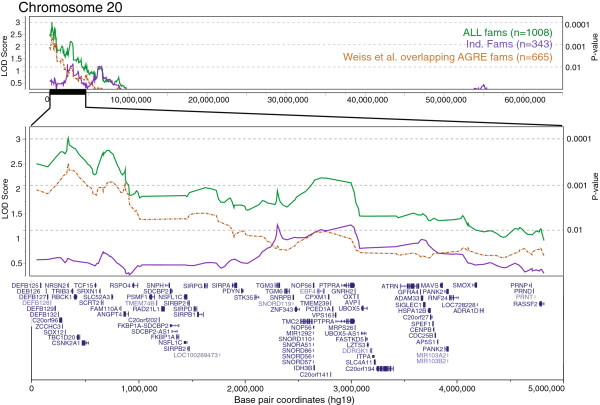
We observed an independent replication of previously observed linkage at chromosome 20p13 (P < 0.01), while loci at 6q27 and 8q13.2 showed suggestive linkage in our extended sample. Suggestive sex-differential linkage was observed at 1p31.3 (MO), 8p21.2 (FC), and 8p12 (FC) in our discovery sample, and the MO signal at 1p31.3 was supported in our expanded sample. No sex-differential signals met replication criteria, and no common SNPs were significantly associated with ASD within any identified linkage regions.
With few exceptions, analyses of subsets of families from the AGRE cohort identify different risk loci, consistent with extreme locus heterogeneity in ASD. Large samples appear to yield more consistent results, and sex-stratified analyses facilitate the identification of sex-differential risk loci, suggesting that linkage analyses in large cohorts are useful for identifying heritable risk loci. Additional work, such as targeted re-sequencing, is needed to identify the specific variants within these loci that are responsible for increasing ASD risk.
自闭症谱系障碍(ASD)是男性偏倚和遗传异质性的。虽然对散发性病例的测序已经确定了新生风险变异,但遗传贡献和驱动男性偏倚的机制仍知之甚少。在这里,我们旨在确定自闭症基因资源交换(AGRE)中最大可用、统一确定、高密度基因分型的多基因 ASD 家族中可遗传和性别差异的风险基因座,并将结果与 AGRE 的早期发现进行比较。
从总共 1008 个多基因家族的样本中,我们在 847 个家族的发现样本中进行了全基因组非参数连锁分析,并分别对仅男性受影响儿童(男性独有的 MO)或至少有一个女性受影响儿童(包含女性的 FC)的家族子集进行了分析。在发现样本中或之前的 AGRE 样本中显示出提示性连锁证据(对数优势≥2.2)的基因座,在利用所有 1008 个可用家族的扩展研究中重新进行了评估。对于在发现阶段具有全基因组显著连锁信号的区域,那些未包含在相应发现样本中的家族则进行了独立复制的连锁评估。还在提示性连锁区域内对常见单核苷酸多态性(SNP)进行了关联测试。
我们观察到先前在 20p13 染色体上观察到的连锁的独立复制(P<0.01),而在我们的扩展样本中,6q27 和 8q13.2 上的基因座显示出提示性连锁。在我们的发现样本中,在 1p31.3(MO)、8p21.2(FC)和 8p12(FC)观察到提示性性别差异连锁,在我们的扩展样本中,MO 信号在 1p31.3 上得到支持。没有性别差异的信号达到复制标准,并且在任何确定的连锁区域内,没有常见的 SNP 与 ASD 显著相关。
除了少数例外,AGRE 队列家族子集的分析确定了不同的风险基因座,这与 ASD 中的极端基因座异质性一致。大样本似乎产生更一致的结果,性别分层分析有助于确定性别差异的风险基因座,这表明在大样本中进行连锁分析对于识别可遗传的风险基因座是有用的。需要进一步的工作,例如靶向重测序,以确定这些基因座内导致 ASD 风险增加的特定变异。