Bu Jingde, Chi Xuebin, Jin Zhong
BMC Syst Biol. 2013;7 Suppl 2(Suppl 2):S10. doi: 10.1186/1752-0509-7-S2-S10. Epub 2013 Dec 17.
RNA-Seq methodology is a revolutionary transcriptomics sequencing technology, which is the representative of Next generation Sequencing (NGS). With the high throughput sequencing of RNA-Seq, we can acquire much more information like differential expression and novel splice variants from deep sequence analysis and data mining. But the short read length brings a great challenge to alignment, especially when the reads span two or more exons.
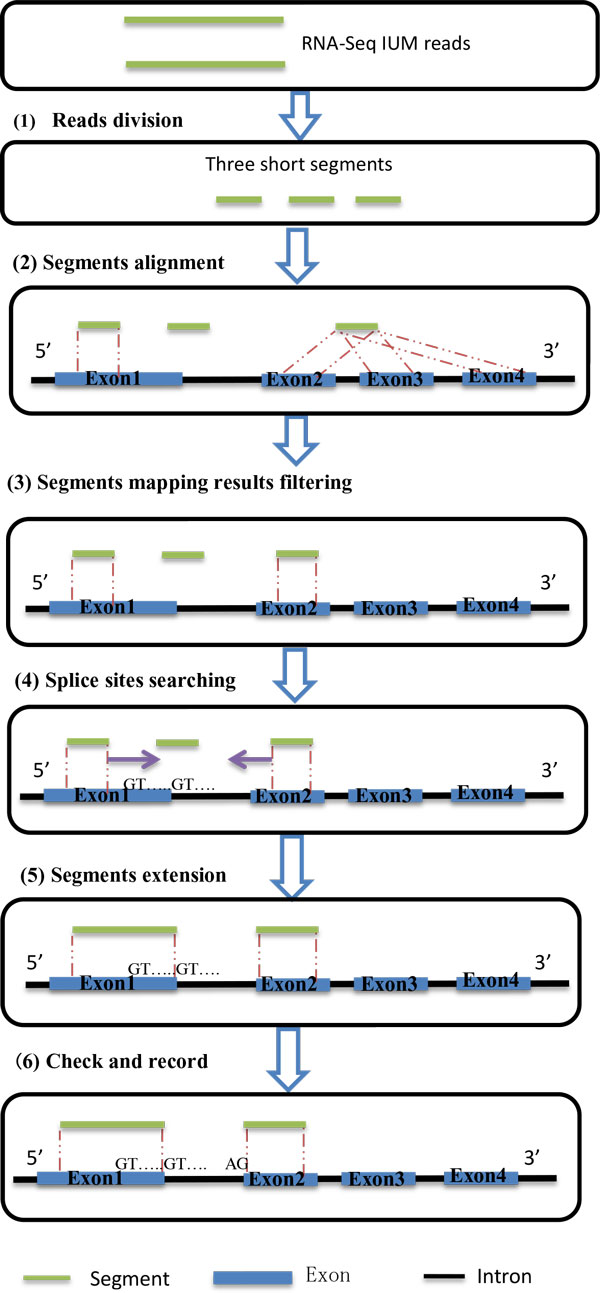
A two steps heuristic splice alignment tool is generated in this investigation. First, map raw reads to reference with unspliced aligner--BWA; second, split initial unmapped reads into three equal short reads (seeds), align each seed to the reference, filter hits, search possible split position of read and extend hits to a complete match.
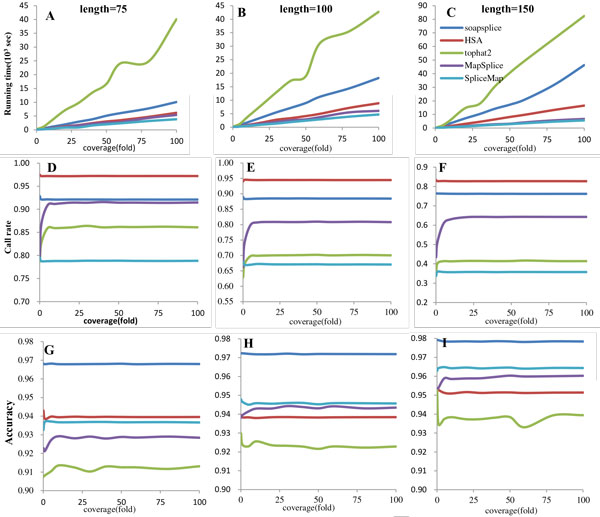
Compare with other splice alignment tools like SOAPsplice and Tophat2, HSA has a better performance in call rate and efficiency, but its results do not as accurate as the other software to some extent.
HSA is an effective spliced aligner of RNA-Seq reads mapping, which is available at https://github.com/vlcc/HSA.
RNA测序方法是一种革命性的转录组测序技术,是新一代测序(NGS)的代表。通过RNA测序的高通量测序,我们可以从深度序列分析和数据挖掘中获得更多信息,如差异表达和新的剪接变体。但是短读长给比对带来了巨大挑战,尤其是当读段跨越两个或更多外显子时。
本研究中生成了一种两步启发式剪接比对工具。首先,使用未剪接比对器BWA将原始读段比对到参考序列;其次,将初始未比对上的读段拆分成三个等长的短读段(种子),将每个种子比对到参考序列,过滤命中结果,搜索读段可能的拆分位置,并将命中结果扩展为完全匹配。
与其他剪接比对工具如SOAPsplice和Tophat2相比,HSA在检出率和效率方面表现更好,但在一定程度上其结果不如其他软件准确。
HSA是一种有效的RNA测序读段映射剪接比对工具,可在https://github.com/vlcc/HSA获取。