Peng Xiaoqing, Wang Jianxin, Zhang Zhen, Xiao Qianghua, Li Min, Pan Yi
BMC Bioinformatics. 2015;16 Suppl 5(Suppl 5):S8. doi: 10.1186/1471-2105-16-S5-S8. Epub 2015 Mar 18.
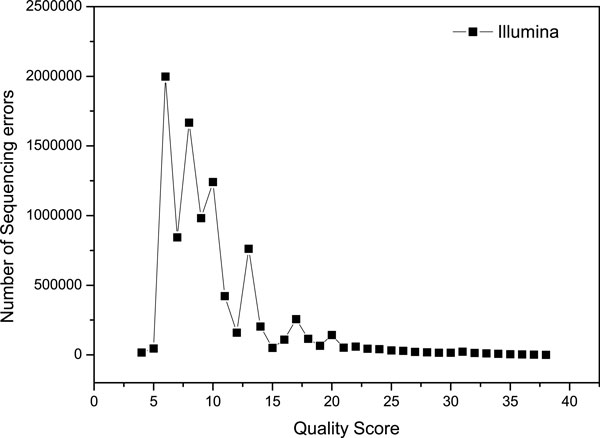
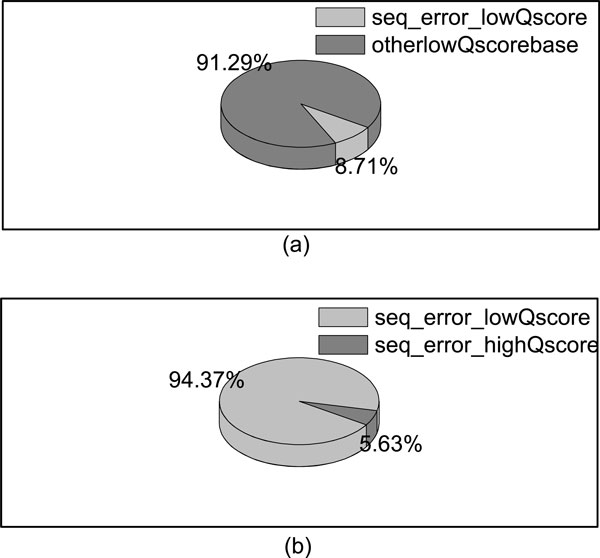
Based on the next generation genome sequencing technologies, a variety of biological applications are developed, while alignment is the first step once the sequencing reads are obtained. In recent years, many software tools have been developed to efficiently and accurately align short reads to the reference genome. However, there are still many reads that can't be mapped to the reference genome, due to the exceeding of allowable mismatches. Moreover, besides the unmapped reads, the reads with low mapping qualities are also excluded from the downstream analysis, such as variance calling. If we can take advantages of the confident segments of these reads, not only can the alignment rates be improved, but also more information will be provided for the downstream analysis.
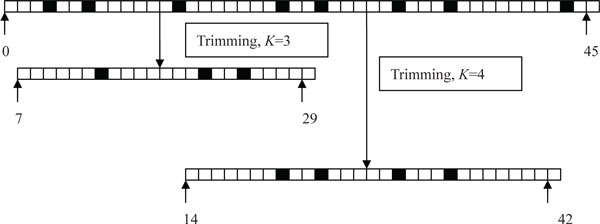
This paper proposes a method, called RAUR (Re-align the Unmapped Reads), to re-align the reads that can not be mapped by alignment tools. Firstly, it takes advantages of the base quality scores (reported by the sequencer) to figure out the most confident and informative segments of the unmapped reads by controlling the number of possible mismatches in the alignment. Then, combined with an alignment tool, RAUR re-align these segments of the reads. We run RAUR on both simulated data and real data with different read lengths. The results show that many reads which fail to be aligned by the most popular alignment tools (BWA and Bowtie2) can be correctly re-aligned by RAUR, with a similar Precision. Even compared with the BWA-MEM and the local mode of Bowtie2, which perform local alignment for long reads to improve the alignment rate, RAUR also shows advantages on the Alignment rate and Precision in some cases. Therefore, the trimming strategy used in RAUR is useful to improve the Alignment rate of alignment tools for the next-generation genome sequencing.
All source code are available at http://netlab.csu.edu.cn/bioinformatics/RAUR.html.
基于下一代基因组测序技术,开发了各种生物学应用,而比对是获得测序读段后的第一步。近年来,已经开发了许多软件工具来高效、准确地将短读段与参考基因组进行比对。然而,由于允许的错配数超标,仍有许多读段无法映射到参考基因组。此外,除了未映射的读段外,映射质量低的读段也被排除在下游分析之外,例如变异检测。如果我们能够利用这些读段的可靠片段,不仅可以提高比对率,还能为下游分析提供更多信息。
本文提出了一种名为RAUR(重新比对未映射读段)的方法,用于重新比对那些无法被比对工具映射的读段。首先,它利用测序仪报告的碱基质量分数,通过控制比对中可能的错配数来找出未映射读段中最可靠和信息量最大的片段。然后,结合一个比对工具,RAUR对这些读段片段进行重新比对。我们在不同读长的模拟数据和真实数据上运行RAUR。结果表明,许多无法被最流行的比对工具(BWA和Bowtie2)比对的读段可以被RAUR正确重新比对,且精度相似。即使与BWA-MEM和Bowtie2的局部模式(对长读段进行局部比对以提高比对率)相比,RAUR在某些情况下在比对率和精度上也具有优势。因此,RAUR中使用的修剪策略有助于提高下一代基因组测序比对工具的比对率。