IntegraGen Evry, France.
Department of Pathology and Laboratory Medicine, University of Pennsylvania Philadelphia, PA, USA.
Front Genet. 2014 Feb 18;5:33. doi: 10.3389/fgene.2014.00033. eCollection 2014.
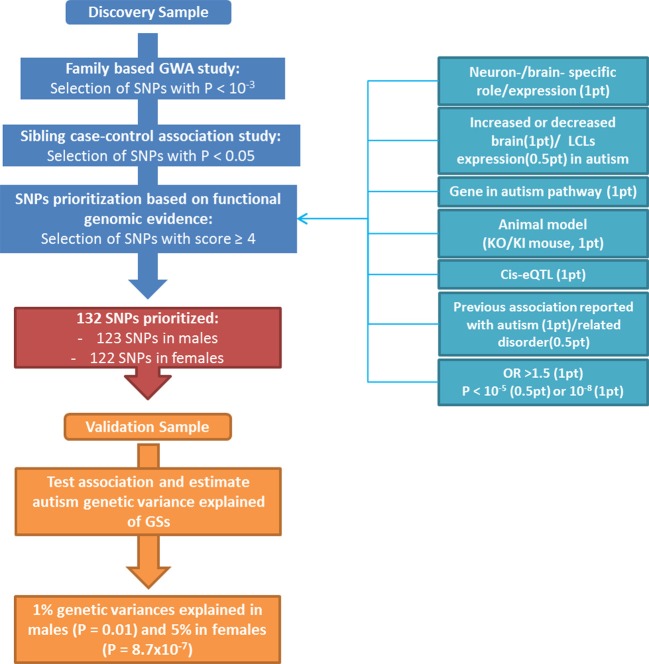
Autism spectrum disorders (ASD) are highly heritable complex neurodevelopmental disorders with a 4:1 male: female ratio. Common genetic variation could explain 40-60% of the variance in liability to autism. Because of their small effect, genome-wide association studies (GWASs) have only identified a small number of individual single-nucleotide polymorphisms (SNPs). To increase the power of GWASs in complex disorders, methods like convergent functional genomics (CFG) have emerged to extract true association signals from noise and to identify and prioritize genes from SNPs using a scoring strategy combining statistics and functional genomics. We adapted and applied this approach to analyze data from a GWAS performed on families with multiple children affected with autism from Autism Speaks Autism Genetic Resource Exchange (AGRE). We identified a set of 133 candidate markers that were localized in or close to genes with functional relevance in ASD from a discovery population (545 multiplex families); a gender specific genetic score (GS) based on these common variants explained 1% (P = 0.01 in males) and 5% (P = 8.7 × 10(-7) in females) of genetic variance in an independent sample of multiplex families. Overall, our work demonstrates that prioritization of GWAS data based on functional genomics identified common variants associated with autism and provided additional support for a common polygenic background in autism.
自闭症谱系障碍(ASD)是一种高度遗传性的复杂神经发育障碍,男女比例为 4:1。常见的遗传变异可以解释自闭症易感性的 40-60%的差异。由于其影响较小,全基因组关联研究(GWAS)仅鉴定出少数个体单核苷酸多态性(SNP)。为了提高 GWAS 在复杂疾病中的效力,像汇聚功能基因组学(CFG)这样的方法已经出现,以从噪声中提取真正的关联信号,并使用结合统计学和功能基因组学的评分策略来识别和优先考虑来自 SNP 的基因。我们对来自自闭症 Speaks 自闭症遗传资源交换(AGRE)的受自闭症影响的多个孩子的家庭进行的 GWAS 数据进行了改编和应用。我们在发现人群(545 个多基因家庭)中鉴定出了一组 133 个候选标记,这些标记位于或靠近与 ASD 具有功能相关性的基因中;基于这些常见变异的性别特异性遗传评分(GS)解释了 1%(男性中 P = 0.01)和 5%(女性中 P = 8.7×10(-7))多基因家庭的遗传方差。总的来说,我们的工作表明,基于功能基因组学对 GWAS 数据进行优先级排序可以识别与自闭症相关的常见变异,并为自闭症的常见多基因背景提供了额外的支持。