Prasad Rathi, Chan Li F, Hughes Claire R, Kaski Juan P, Kowalczyk Julia C, Savage Martin O, Peters Catherine J, Nathwani Nisha, Clark Adrian J L, Storr Helen L, Metherell Louise A
Centre for Endocrinology (R.P., L.F.C., C.R.H., J.C.K., M.O.S., A.J.L.C., H.L.S., L.A.M.), Queen Mary University of London, William Harvey Research Institute, Barts and the London School of Medicine and Dentistry, London EC1M 6BQ, United Kingdom; Inherited Cardiovascular Diseases Unit (J.P.K.), Department of Cardiology, Great Ormond St Hospital for Children, and Department of Paediatric Endocrinology (C.J.P.), Great Ormond St Hospital for Children, London WC1N 3JH, United Kingdom; and Department of Paediatric Endocrinology (N.N.), Luton and Dunstable University Hospital, Luton LU4 0DZ, United Kingdom.
J Clin Endocrinol Metab. 2014 Aug;99(8):E1556-63. doi: 10.1210/jc.2013-3844. Epub 2014 Mar 6.
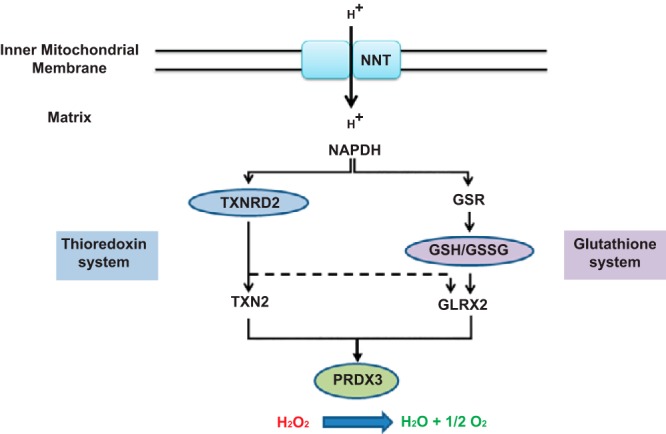
Classic ACTH resistance, due to disruption of ACTH signaling, accounts for the majority of cases of familial glucocorticoid deficiency (FGD). Recently FGD cases caused by mutations in the mitochondrial antioxidant, nicotinamide nucleotide transhydrogenase, have highlighted the importance of redox regulation in steroidogenesis.
We hypothesized that other components of mitochondrial antioxidant systems would be good candidates in the etiology of FGD.
Whole-exome sequencing was performed on three related patients, and segregation of putative causal variants confirmed by Sanger sequencing of all family members. A TXNRD2-knockdown H295R cell line was created to investigate redox homeostasis.
The study was conducted on patients from three pediatric centers in the United Kingdom.
Seven individuals from a consanguineous Kashmiri kindred, six of whom presented with FGD between 0.1 and 10.8 years, participated in the study.
There were no interventions.
Identification and functional interrogation of a novel homozygous mutation segregating with the disease trait were measured.
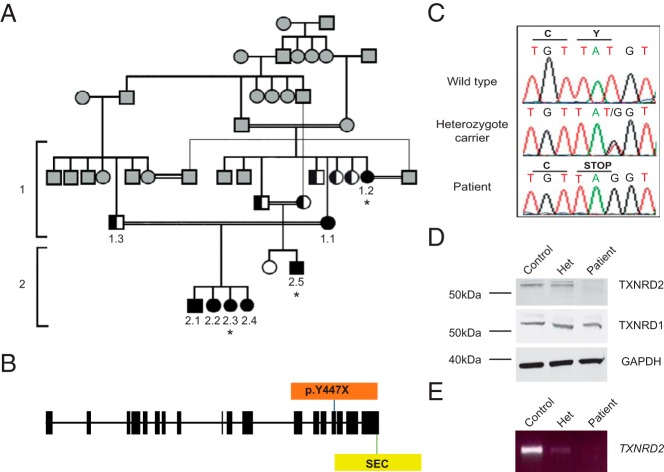
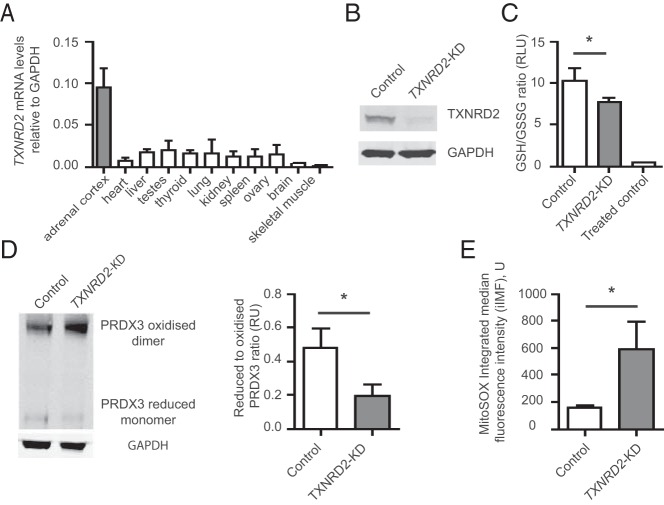
A stop gain mutation, p.Y447X in TXNRD2, encoding the mitochondrial selenoprotein thioredoxin reductase 2 (TXNRD2) was identified and segregated with disease in this extended kindred. RT-PCR and Western blotting revealed complete absence of TXNRD2 in patients homozygous for the mutation. TXNRD2 deficiency leads to impaired redox homeostasis in a human adrenocortical cell line.
In contrast to the Txnrd2-knockout mouse model, in which embryonic lethality as a consequence of hematopoietic and cardiac defects is described, absence of TXNRD2 in humans leads to glucocorticoid deficiency. This is the first report of a homozygous mutation in any component of the thioredoxin antioxidant system leading to inherited disease in humans.
经典促肾上腺皮质激素(ACTH)抵抗是由于ACTH信号传导中断引起的,占家族性糖皮质激素缺乏症(FGD)病例的大多数。最近,由线粒体抗氧化剂烟酰胺核苷酸转氢酶突变引起的FGD病例突出了氧化还原调节在类固醇生成中的重要性。
我们假设线粒体抗氧化系统的其他成分可能是FGD病因的良好候选因素。
对三名相关患者进行了全外显子组测序,并通过对所有家庭成员的桑格测序确认了推定的致病变异的分离情况。创建了TXNRD2基因敲低的H295R细胞系以研究氧化还原稳态。
该研究在英国三个儿科中心的患者中进行。
来自克什米尔近亲家族的七个人参与了研究,其中六人在0.1至10.8岁之间出现FGD。
无干预措施。
测量与疾病特征相关的新型纯合突变的鉴定和功能研究。
在这个大家族中,鉴定出编码线粒体硒蛋白硫氧还蛋白还原酶2(TXNRD2)的TXNRD2基因中的一个截短突变p.Y447X,并与疾病分离。逆转录聚合酶链反应(RT-PCR)和蛋白质免疫印迹显示,该突变纯合患者中完全不存在TXNRD2。TXNRD2缺乏导致人肾上腺皮质细胞系中的氧化还原稳态受损。
与Txnrd2基因敲除小鼠模型不同,后者描述了由于造血和心脏缺陷导致的胚胎致死性,而人类中TXNRD2的缺失会导致糖皮质激素缺乏。这是硫氧还蛋白抗氧化系统任何成分中的纯合突变导致人类遗传性疾病的首次报道。