Department of Internal Medicine, The University of Texas Medical Branch, Galveston, TX 775555, USA.
Department of Pathology, The University of Texas Medical Branch, Galveston, TX 775555, USA.
Toxicol Appl Pharmacol. 2014 Jun 1;277(2):109-17. doi: 10.1016/j.taap.2014.02.018. Epub 2014 Mar 10.
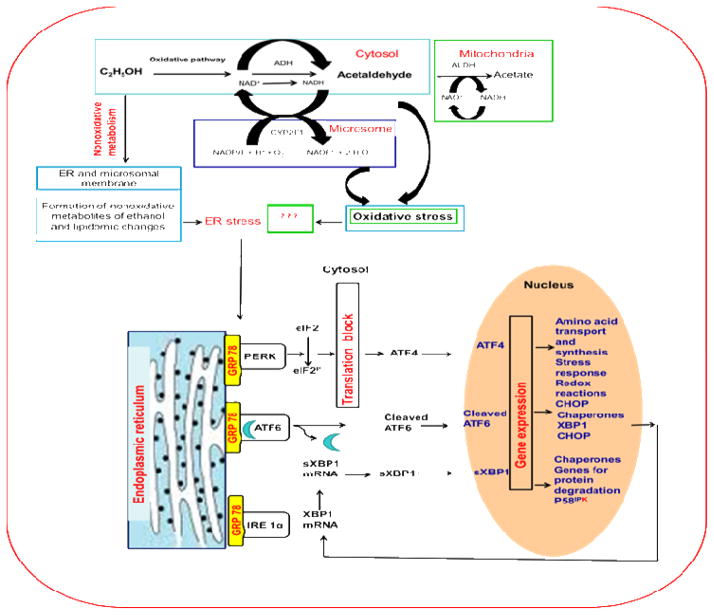
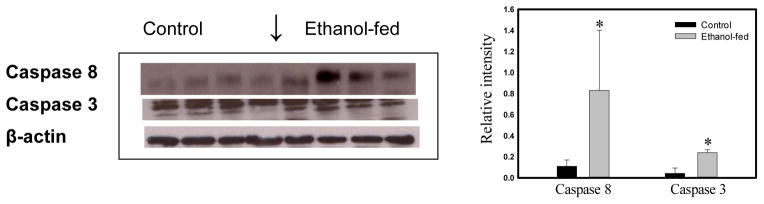
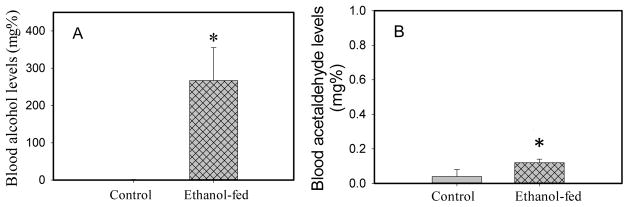
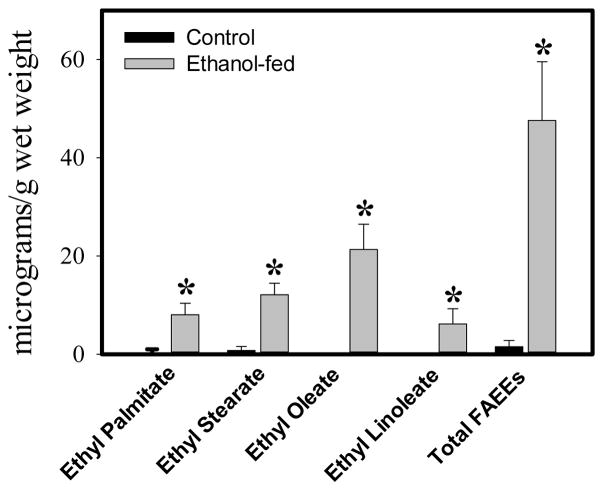
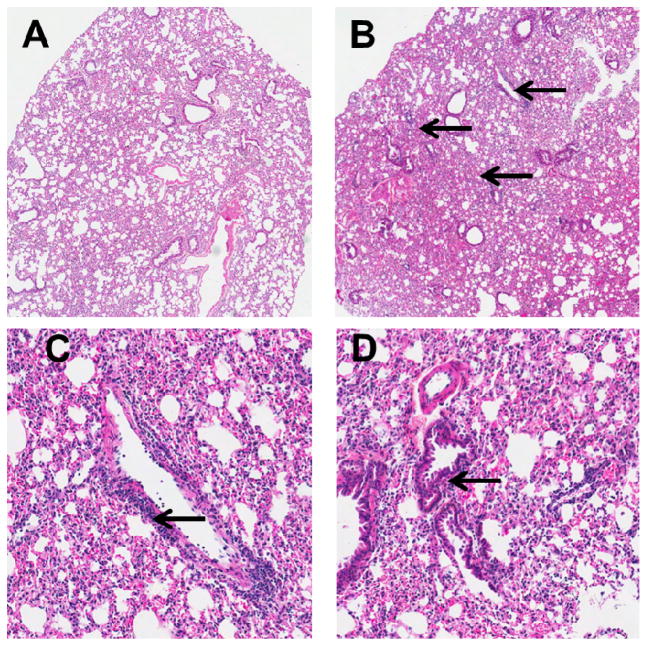
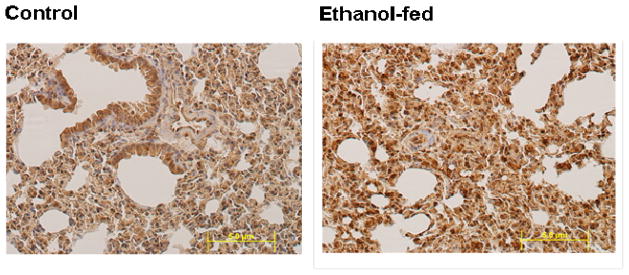
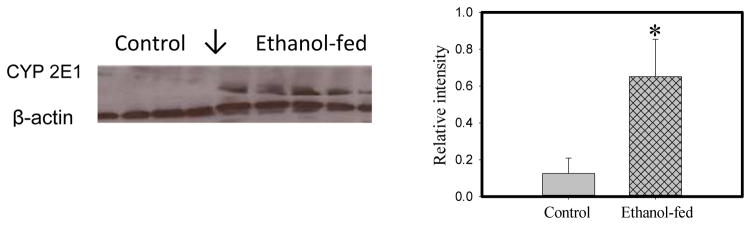
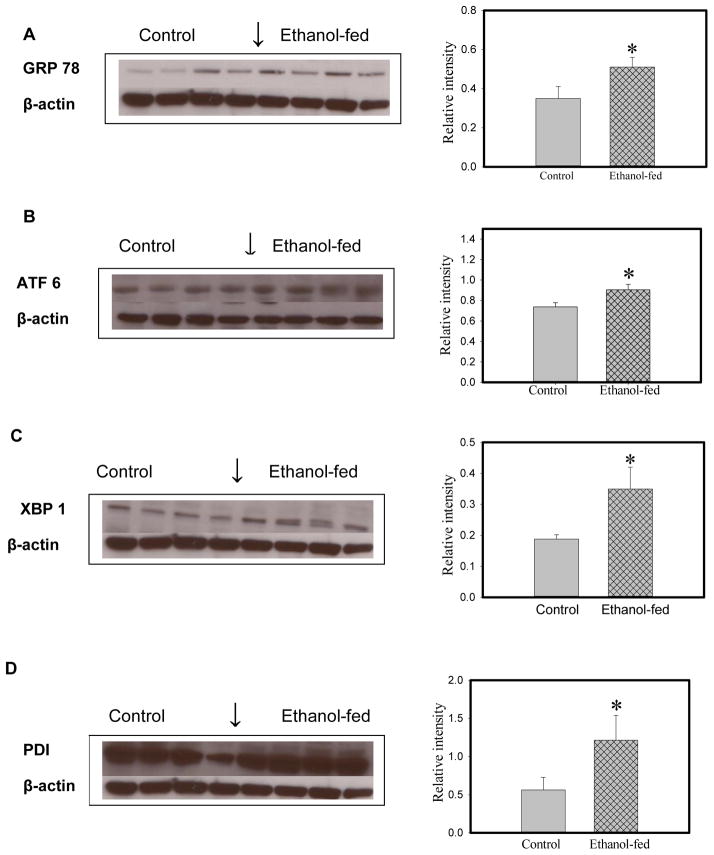
Consumption and over-consumption of alcoholic beverages are well-recognized contributors to a variety of pulmonary disorders, even in the absence of intoxication. The mechanisms by which alcohol (ethanol) may produce disease include oxidative stress and prolonged endoplasmic reticulum (ER) stress. Many aspects of these processes remain incompletely understood due to a lack of a suitable animal model. Chronic alcohol over-consumption reduces hepatic alcohol dehydrogenase (ADH), the principal canonical metabolic pathway of ethanol oxidation. We therefore modeled this situation using hepatic ADH-deficient deer mice fed 3.5% ethanol daily for 3 months. Blood ethanol concentration was 180 mg% in ethanol fed mice, compared to <1.0% in the controls. Acetaldehyde (oxidative metabolite of ethanol) was minimally, but significantly increased in ethanol-fed vs. pair-fed control mice. Total fatty acid ethyl esters (FAEEs, nonoxidative metabolites of ethanol) were 47.6 μg/g in the lungs of ethanol-fed mice as compared to 1.5 μg/g in pair-fed controls. Histological and immunohistological evaluation showed perivascular and peribronchiolar lymphocytic infiltration, and significant oxidative injury, in the lungs of ethanol-fed mice compared to pair-fed controls. Several fold increases for cytochrome P450 2E1, caspase 8 and caspase 3 found in the lungs of ethanol-fed mice as compared to pair-fed controls suggest role of oxidative stress in ethanol-induced lung injury. ER stress and unfolded protein response signaling were also significantly increased in the lungs of ethanol-fed mice. Surprisingly, no significant activation of inositol-requiring enzyme-1α and spliced XBP1 was observed indicating a lack of activation of corrective mechanisms to reinstate ER homeostasis. The data suggest that oxidative stress and prolonged ER stress, coupled with formation and accumulation of cytotoxic FAEEs may contribute to the pathogenesis of alcoholic lung disease.
饮酒和酗酒是多种肺部疾病的公认病因,即使在没有中毒的情况下也是如此。酒精(乙醇)可能导致疾病的机制包括氧化应激和内质网(ER)应激的延长。由于缺乏合适的动物模型,这些过程的许多方面仍然不完全了解。慢性酒精过度消耗会降低肝脏中的酒精脱氢酶(ADH),这是乙醇氧化的主要经典代谢途径。因此,我们使用肝脏 ADH 缺乏的鹿鼠模型来模拟这种情况,这些鹿鼠每天喂食 3.5%的乙醇,持续 3 个月。与对照组相比,喂食乙醇的小鼠血液中的乙醇浓度为 180mg%,而对照组中<1.0%。与对照组相比,喂食乙醇的小鼠中乙醛(乙醇的氧化代谢物)略有但显著增加。与对照组相比,喂食乙醇的小鼠肺部的总脂肪酸乙酯(乙醇的非氧化代谢物)为 47.6μg/g,而对照组为 1.5μg/g。与对照组相比,喂食乙醇的小鼠肺部的血管周围和细支气管周围淋巴细胞浸润和明显的氧化损伤明显增加。与对照组相比,喂食乙醇的小鼠肺部的细胞色素 P450 2E1、半胱天冬酶 8 和半胱天冬酶 3 增加了几倍,表明氧化应激在乙醇诱导的肺损伤中起作用。内质网应激和未折叠蛋白反应信号也显著增加。令人惊讶的是,与对照组相比,未观察到肌醇需求酶 1α和剪接 XBP1 的明显激活,这表明纠正内质网稳态的修复机制没有激活。数据表明,氧化应激和延长的内质网应激,加上细胞毒性 FAEE 的形成和积累,可能导致酒精性肺病的发病机制。