Boley Nathan, Stoiber Marcus H, Booth Benjamin W, Wan Kenneth H, Hoskins Roger A, Bickel Peter J, Celniker Susan E, Brown James B
Department of Biostatistics, University of California at Berkeley, Berkeley, California, USA.
Department of Genome Dynamics, Lawrence Berkeley National Laboratory, Berkeley, California, USA.
Nat Biotechnol. 2014 Apr;32(4):341-6. doi: 10.1038/nbt.2850. Epub 2014 Mar 16.
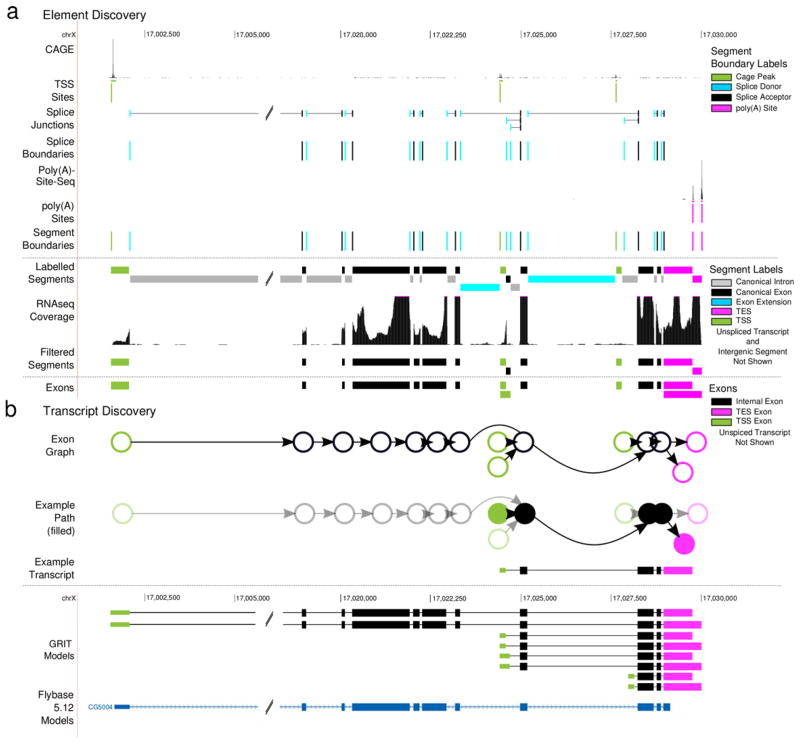
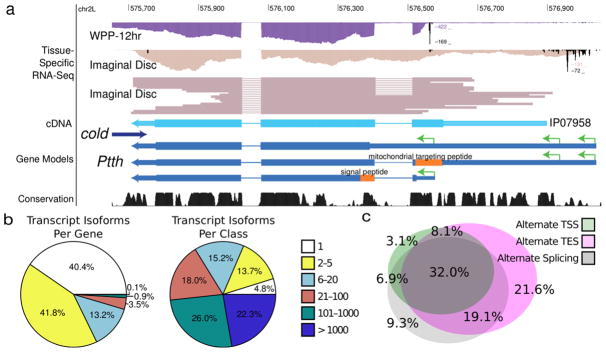
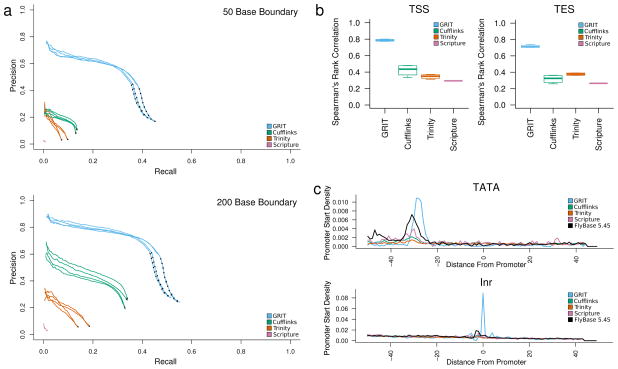
The identification of full length transcripts entirely from short-read RNA sequencing data (RNA-seq) remains a challenge in the annotation of genomes. Here we describe an automated pipeline for genome annotation that integrates RNA-seq and gene-boundary data sets, which we call Generalized RNA Integration Tool, or GRIT. Applying GRIT to Drosophila melanogaster short-read RNA-seq, cap analysis of gene expression (CAGE) and poly(A)-site-seq data collected for the modENCODE project, we recovered the vast majority of previously annotated transcripts and doubled the total number of transcripts cataloged. We found that 20% of protein coding genes encode multiple protein-localization signals and that, in 20-d-old adult fly heads, genes with multiple polyadenylation sites are more common than genes with alternative splicing or alternative promoters. GRIT demonstrates 30% higher precision and recall than the most widely used transcript assembly tools. GRIT will facilitate the automated generation of high-quality genome annotations without the need for extensive manual annotation.
仅从短读长RNA测序数据(RNA-seq)中识别全长转录本,仍然是基因组注释中的一项挑战。在此,我们描述了一种用于基因组注释的自动化流程,该流程整合了RNA-seq和基因边界数据集,我们将其称为通用RNA整合工具(Generalized RNA Integration Tool,简称GRIT)。将GRIT应用于为modENCODE项目收集的黑腹果蝇短读长RNA-seq、基因表达的帽分析(CAGE)和聚腺苷酸化位点测序数据,我们找回了绝大多数先前注释的转录本,并使编目转录本的总数增加了一倍。我们发现,20%的蛋白质编码基因编码多个蛋白质定位信号,并且在20日龄成年果蝇头部,具有多个聚腺苷酸化位点的基因比具有可变剪接或可变启动子的基因更常见。GRIT的精确率和召回率比使用最广泛的转录本组装工具高30%。GRIT将有助于自动生成高质量的基因组注释,而无需大量人工注释。