Division of Biology, California Institute of Technology, Pasadena, California 91125, USA.
Genome Res. 2012 Oct;22(10):2079-87. doi: 10.1101/gr.139170.112. Epub 2012 Jun 18.
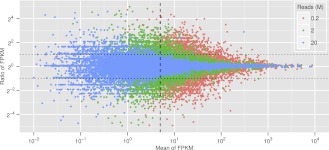
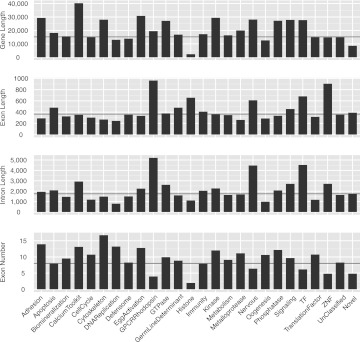
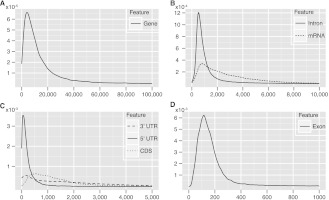
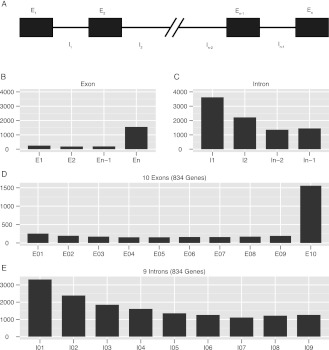
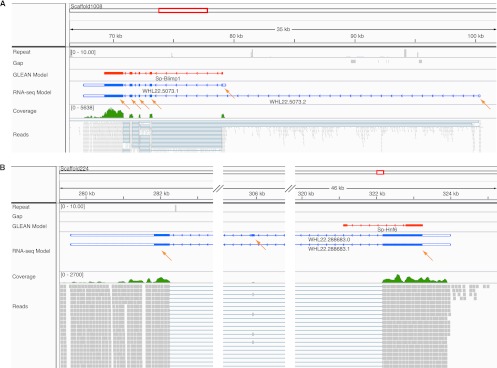
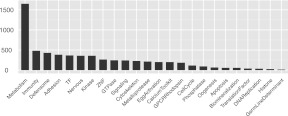
A comprehensive transcriptome analysis has been performed on protein-coding RNAs of Strongylocentrotus purpuratus, including 10 different embryonic stages, six feeding larval and metamorphosed juvenile stages, and six adult tissues. In this study, we pooled the transcriptomes from all of these sources and focused on the insights they provide for gene structure in the genome of this recently sequenced model system. The genome had initially been annotated by use of computational gene model prediction algorithms. A large fraction of these predicted genes were recovered in the transcriptome when the reads were mapped to the genome and appropriately filtered and analyzed. However, in a manually curated subset, we discovered that more than half the computational gene model predictions were imperfect, containing errors such as missing exons, prediction of nonexistent exons, erroneous intron/exon boundaries, fusion of adjacent genes, and prediction of multiple genes from single genes. The transcriptome data have been used to provide a systematic upgrade of the gene model predictions throughout the genome, very greatly improving the research usability of the genomic sequence. We have constructed new public databases that incorporate information from the transcriptome analyses. The transcript-based gene model data were used to define average structural parameters for S. purpuratus protein-coding genes. In addition, we constructed a custom sea urchin gene ontology, and assigned about 7000 different annotated transcripts to 24 functional classes. Strong correlations became evident between given functional ontology classes and structural properties, including gene size, exon number, and exon and intron size.
已对扁形动物盘虫的蛋白质编码 RNA 进行了全面的转录组分析,包括 10 个不同的胚胎阶段、6 个摄食幼虫和变态后的幼体阶段以及 6 个成年组织。在这项研究中,我们汇集了所有这些来源的转录组,并重点关注它们为这个最近测序的模型系统的基因组中基因结构提供的见解。最初,该基因组是使用计算基因模型预测算法进行注释的。当将读取映射到基因组并进行适当的过滤和分析时,这些预测基因中有很大一部分可以在转录组中恢复。然而,在经过人工编辑的子集,我们发现,超过一半的计算基因模型预测并不完善,包含错误,如缺失外显子、预测不存在的外显子、错误的内含子/外显子边界、相邻基因融合以及从单个基因预测多个基因。转录组数据已被用于提供整个基因组中基因模型预测的系统升级,极大地提高了基因组序列的研究可用性。我们构建了新的公共数据库,这些数据库整合了转录组分析的信息。基于转录本的基因模型数据用于定义扁形动物盘虫蛋白质编码基因的平均结构参数。此外,我们构建了一个定制的海胆基因本体论,并将大约 7000 个不同注释的转录本分配到 24 个功能类别中。在给定的功能本体论类别和结构特性之间,包括基因大小、外显子数量以及外显子和内含子大小之间,出现了明显的相关性。