Faculty of Infectious and Tropical Diseases, London School of Hygiene & Tropical Medicine, WC1E 7HT London, UK.
Faculty of Infectious and Tropical Diseases, London School of Hygiene & Tropical Medicine, WC1E 7HT London, UK.
Tuberculosis (Edinb). 2014 May;94(3):346-54. doi: 10.1016/j.tube.2014.02.005. Epub 2014 Feb 15.
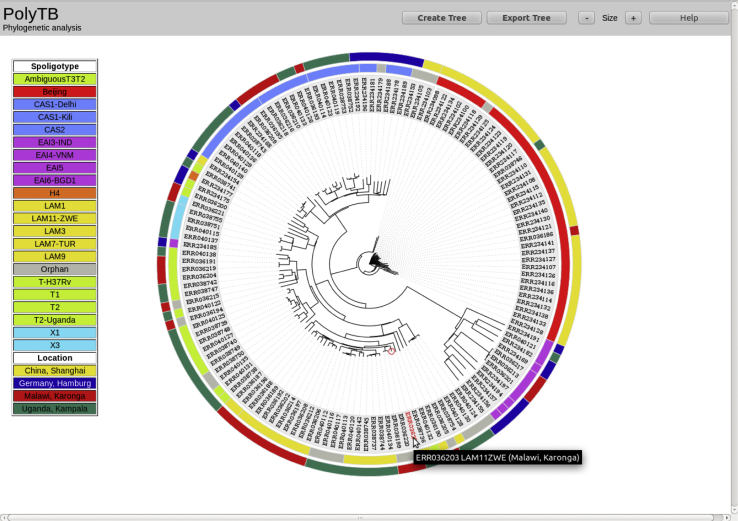
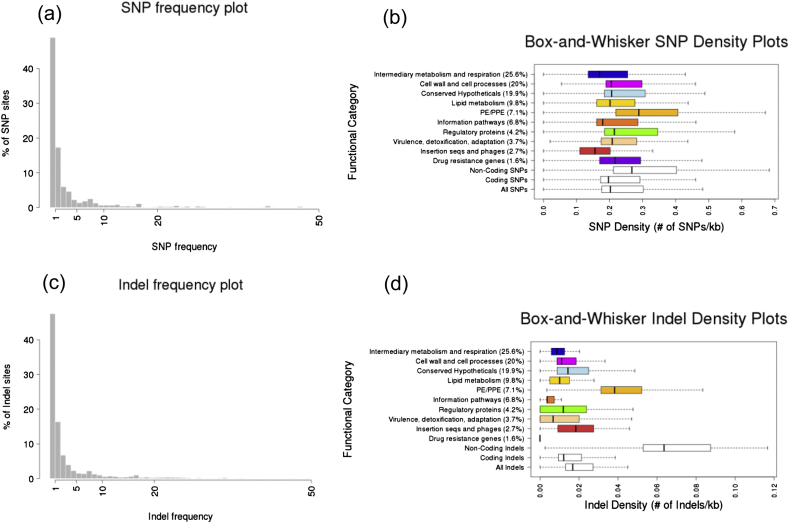
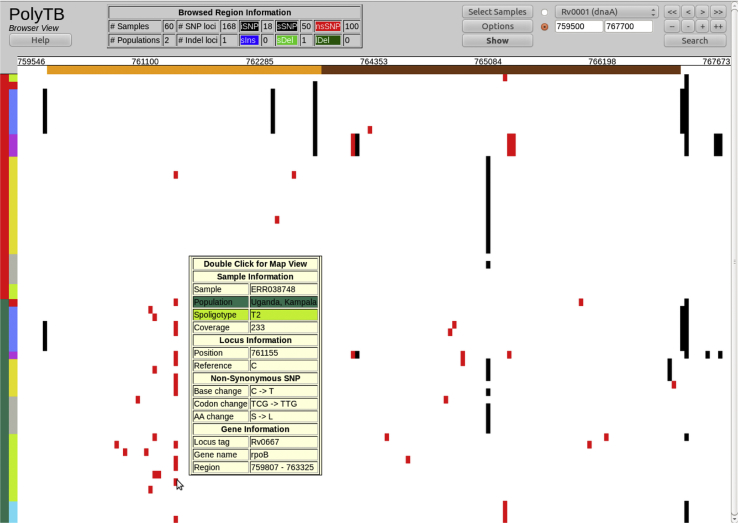
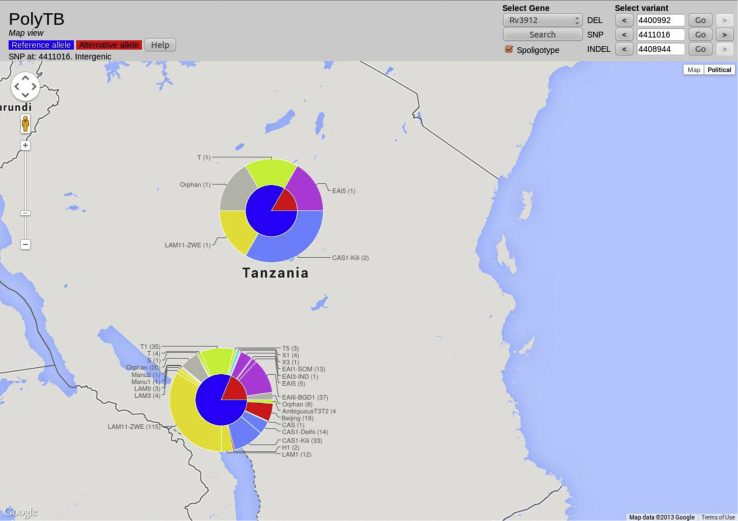
Tuberculosis (TB) caused by Mycobacterium tuberculosis (Mtb) is the second major cause of death from an infectious disease worldwide. Recent advances in DNA sequencing are leading to the ability to generate whole genome information in clinical isolates of M. tuberculosis complex (MTBC). The identification of informative genetic variants such as phylogenetic markers and those associated with drug resistance or virulence will help barcode Mtb in the context of epidemiological, diagnostic and clinical studies. Mtb genomic datasets are increasingly available as raw sequences, which are potentially difficult and computer intensive to process, and compare across studies. Here we have processed the raw sequence data (>1500 isolates, eight studies) to compile a catalogue of SNPs (n = 74,039, 63% non-synonymous, 51.1% in more than one isolate, i.e. non-private), small indels (n = 4810) and larger structural variants (n = 800). We have developed the PolyTB web-based tool (http://pathogenseq.lshtm.ac.uk/polytb) to visualise the resulting variation and important meta-data (e.g. in silico inferred strain-types, location) within geographical map and phylogenetic views. This resource will allow researchers to identify polymorphisms within candidate genes of interest, as well as examine the genomic diversity and distribution of strains. PolyTB source code is freely available to researchers wishing to develop similar tools for their pathogen of interest.
结核分枝杆菌(Mycobacterium tuberculosis,Mtb)引起的结核病是全球第二大致死传染病。最近 DNA 测序技术的进步使人们能够从结核分枝杆菌复合群(Mycobacterium tuberculosis complex,MTBC)的临床分离株中生成全基因组信息。鉴定具有信息性的遗传变异,如系统发育标记和与耐药性或毒力相关的变异,将有助于在流行病学、诊断和临床研究中对 Mtb 进行标记。Mtb 基因组数据集越来越多地以原始序列的形式提供,这些序列在处理和比较方面具有潜在的困难和计算机密集性。在这里,我们处理了原始序列数据(>1500 个分离株,8 项研究),编制了一个 SNP 目录(n=74039,63%是非同义的,51.1%在不止一个分离株中,即非私有)、小插入缺失(n=4810)和较大的结构变异(n=800)。我们开发了基于网络的 PolyTB 工具(http://pathogenseq.lshtm.ac.uk/polytb),以便在地理地图和系统发育视图中可视化由此产生的变异和重要元数据(例如,基于计算机推断的菌株类型、位置)。该资源将允许研究人员识别候选基因中的多态性,并检查菌株的基因组多样性和分布。PolyTB 的源代码可供希望为其感兴趣的病原体开发类似工具的研究人员免费使用。