Bioinformatics Group, Department of Computer Science and the Interdisciplinary Center for Bioinformatic, University of Leipzig, Härtelstraße 16-18, 04107 Leipzig, Germany.
BMC Bioinformatics. 2014 Mar 27;15:89. doi: 10.1186/1471-2105-15-89.
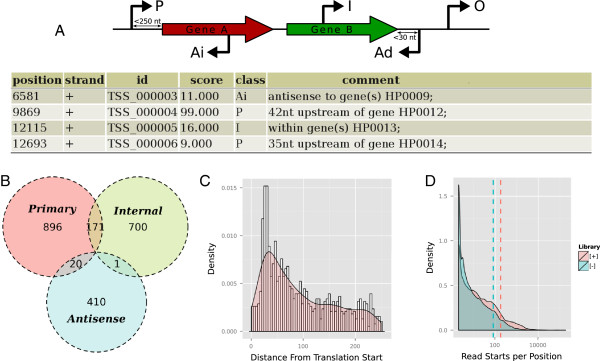
Differential RNA sequencing (dRNA-seq) is a high-throughput screening technique designed to examine the architecture of bacterial operons in general and the precise position of transcription start sites (TSS) in particular. Hitherto, dRNA-seq data were analyzed by visualizing the sequencing reads mapped to the reference genome and manually annotating reliable positions. This is very labor intensive and, due to the subjectivity, biased.
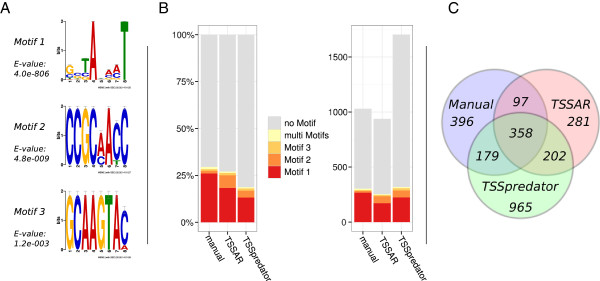
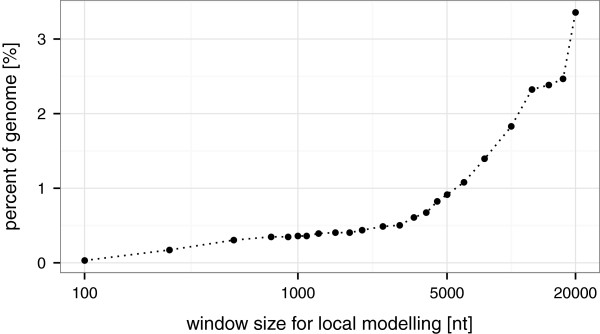
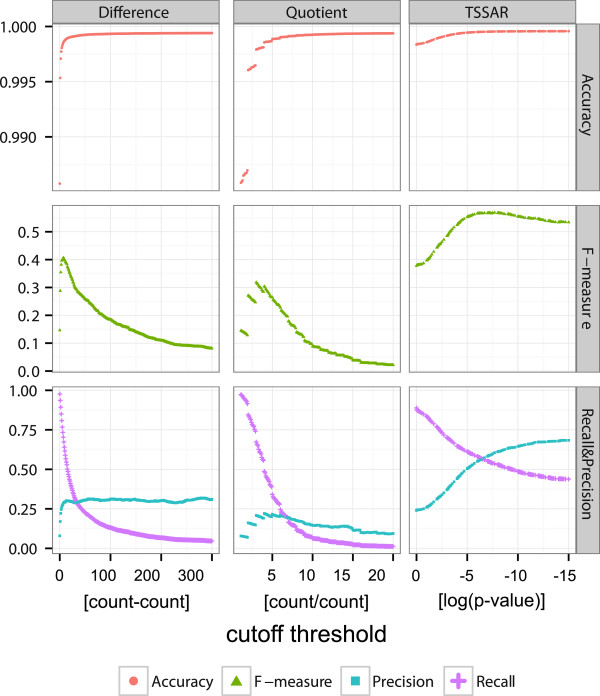
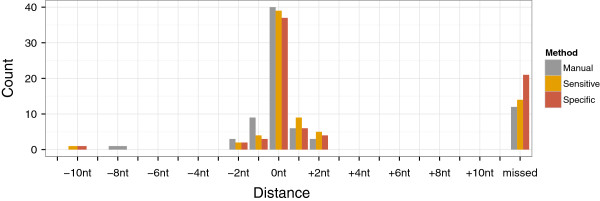
Here, we present TSSAR, a tool for automated de novo TSS annotation from dRNA-seq data that respects the statistics of dRNA-seq libraries. TSSAR uses the premise that the number of sequencing reads starting at a certain genomic position within a transcriptional active region follows a Poisson distribution with a parameter that depends on the local strength of expression. The differences of two dRNA-seq library counts thus follow a Skellam distribution. This provides a statistical basis to identify significantly enriched primary transcripts.We assessed the performance by analyzing a publicly available dRNA-seq data set using TSSAR and two simple approaches that utilize user-defined score cutoffs. We evaluated the power of reproducing the manual TSS annotation. Furthermore, the same data set was used to reproduce 74 experimentally validated TSS in H. pylori from reliable techniques such as RACE or primer extension. Both analyses showed that TSSAR outperforms the static cutoff-dependent approaches.
Having an automated and efficient tool for analyzing dRNA-seq data facilitates the use of the dRNA-seq technique and promotes its application to more sophisticated analysis. For instance, monitoring the plasticity and dynamics of the transcriptomal architecture triggered by different stimuli and growth conditions becomes possible.The main asset of a novel tool for dRNA-seq analysis that reaches out to a broad user community is usability. As such, we provide TSSAR both as intuitive RESTful Web service ( http://rna.tbi.univie.ac.at/TSSAR) together with a set of post-processing and analysis tools, as well as a stand-alone version for use in high-throughput dRNA-seq data analysis pipelines.
差异 RNA 测序(dRNA-seq)是一种高通量筛选技术,旨在研究细菌操纵子的结构,特别是转录起始位点(TSS)的精确位置。迄今为止,dRNA-seq 数据的分析方法是通过可视化映射到参考基因组的测序读数,并手动注释可靠的位置。这非常耗费人力,并且由于主观性,存在偏差。
在这里,我们提出了 TSSAR,这是一种从 dRNA-seq 数据中自动注释从头 TSS 的工具,它尊重 dRNA-seq 文库的统计信息。TSSAR 的前提是,在转录活跃区域内的某个基因组位置开始的测序读数数量遵循泊松分布,其参数取决于局部表达强度。因此,两个 dRNA-seq 文库计数的差异遵循斯凯利分布。这为识别显著富集的初级转录本提供了统计依据。我们通过使用 TSSAR 和两种简单的方法(利用用户定义的评分阈值)分析了一个公开的 dRNA-seq 数据集来评估性能。我们评估了重现手动 TSS 注释的能力。此外,同一数据集用于重现 74 个在 H. pylori 中通过可靠技术(如 RACE 或引物延伸)验证的 TSS。这两项分析都表明,TSSAR 优于基于静态阈值的方法。
拥有一种用于分析 dRNA-seq 数据的自动化和高效工具,便于使用 dRNA-seq 技术,并促进其在更复杂的分析中的应用。例如,通过不同的刺激和生长条件监测转录组结构的可塑性和动态性成为可能。一种新型的 dRNA-seq 分析工具的主要资产是易用性。因此,我们提供了 TSSAR,它既是直观的基于 REST 的 Web 服务(http://rna.tbi.univie.ac.at/TSSAR),也提供了一组后处理和分析工具,以及一个独立的版本,用于高通量 dRNA-seq 数据分析管道。