Genome Sequencing and Analysis Program, The Broad Institute of MIT and Harvard, Cambridge, MA 02141, USA.
Genome Biol. 2012;13(3):R23. doi: 10.1186/gb-2012-13-3-r23.
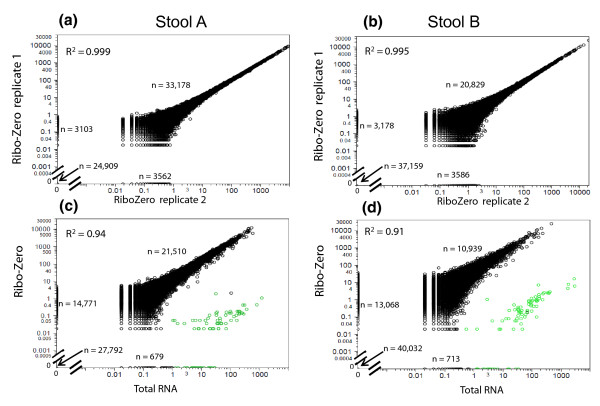
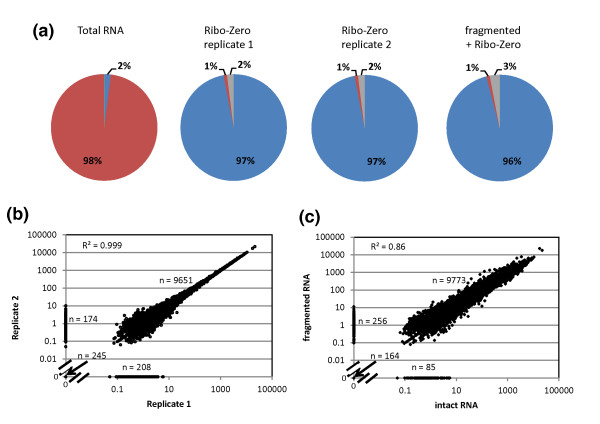
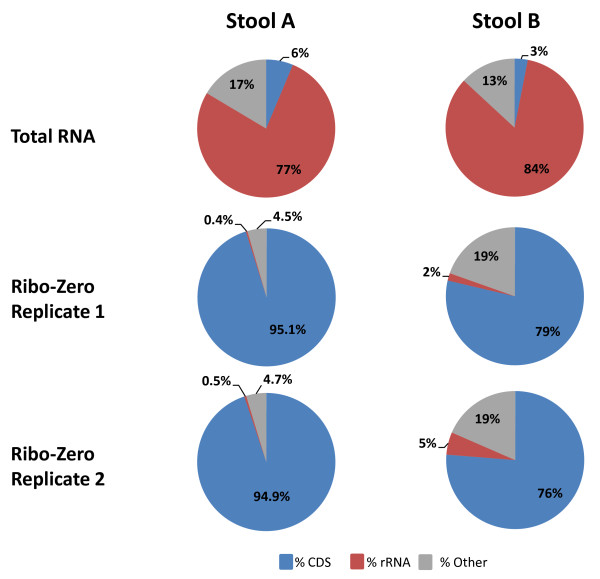
We have developed a process for transcriptome analysis of bacterial communities that accommodates both intact and fragmented starting RNA and combines efficient rRNA removal with strand-specific RNA-seq. We applied this approach to an RNA mixture derived from three diverse cultured bacterial species and to RNA isolated from clinical stool samples. The resulting expression profiles were highly reproducible, enriched up to 40-fold for non-rRNA transcripts, and correlated well with profiles representing undepleted total RNA.
我们开发了一种用于细菌群落转录组分析的方法,该方法既适用于完整的和碎片化的起始 RNA,又能结合高效的 rRNA 去除和链特异性 RNA-seq。我们将该方法应用于源自三种不同培养细菌物种的 RNA 混合物和从临床粪便样本中分离的 RNA。得到的表达谱具有高度的可重复性,非 rRNA 转录本富集高达 40 倍,并且与代表未耗尽总 RNA 的谱很好地相关。