Zhang J-S, Herreros-Villanueva M, Koenig A, Deng Z, de Narvajas A A-M, Gomez T S, Meng X, Bujanda L, Ellenrieder V, Li X K, Kaufmann S H, Billadeau D D
1] Division of Oncology Research and Schulze Center for Novel Therapeutics, Mayo Clinic College of Medicine, Rochester, MN, USA [2] School of Pharmaceutical Sciences and Key Laboratory of Biotechnology and Pharmaceutical Engineering, Wenzhou Medical University, Wenzhou, Zhejiang, PR China.
1] Division of Oncology Research and Schulze Center for Novel Therapeutics, Mayo Clinic College of Medicine, Rochester, MN, USA [2] Department of Gastroenterology and Endocrinology, Philipps University of Marburg, Marburg, Germany.
Cell Death Dis. 2014 Mar 27;5(3):e1142. doi: 10.1038/cddis.2014.102.
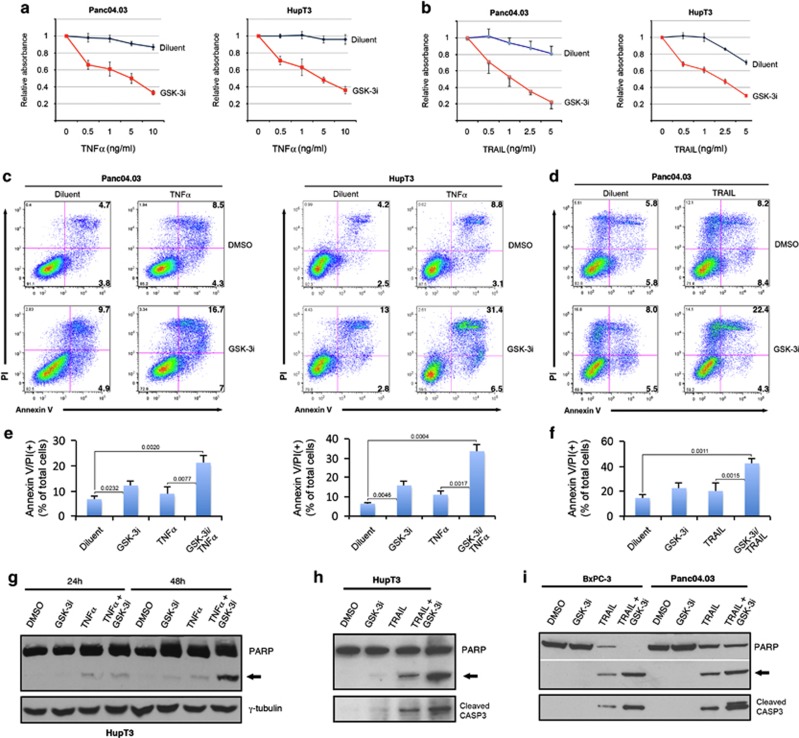
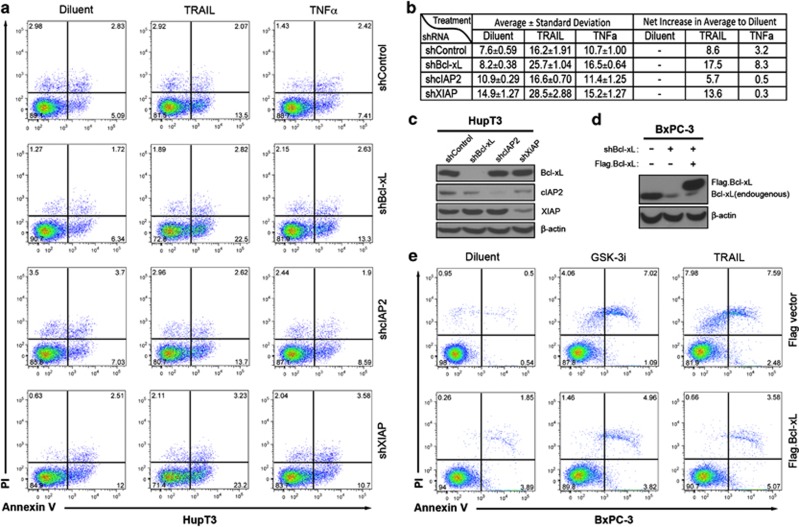
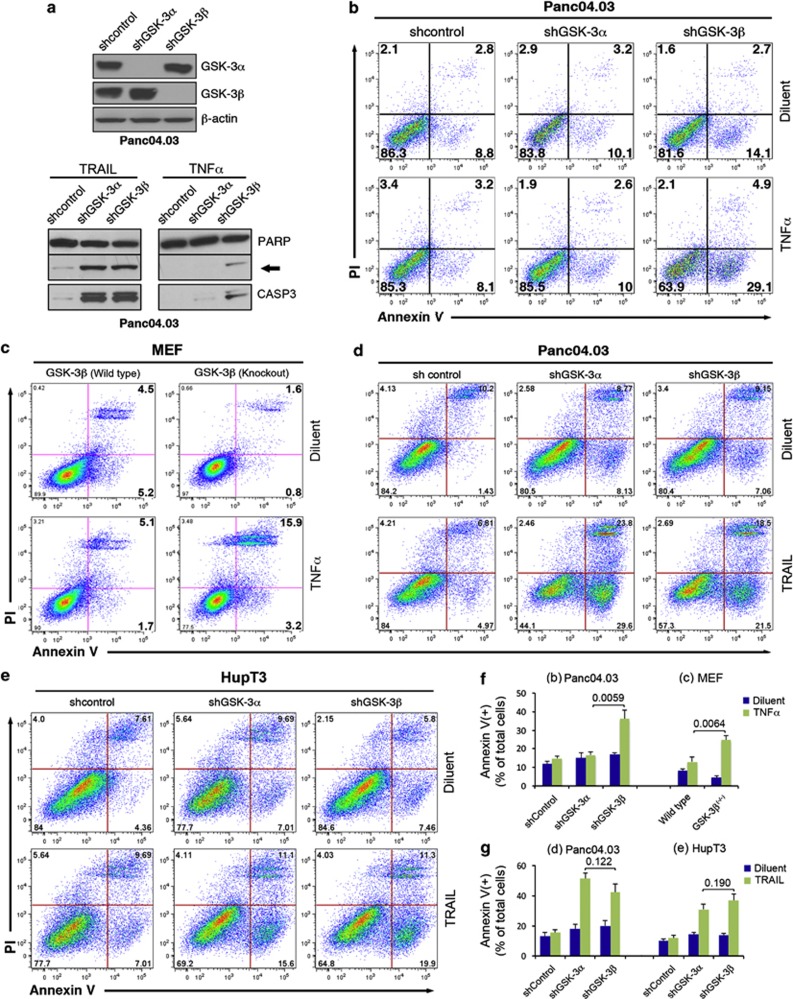
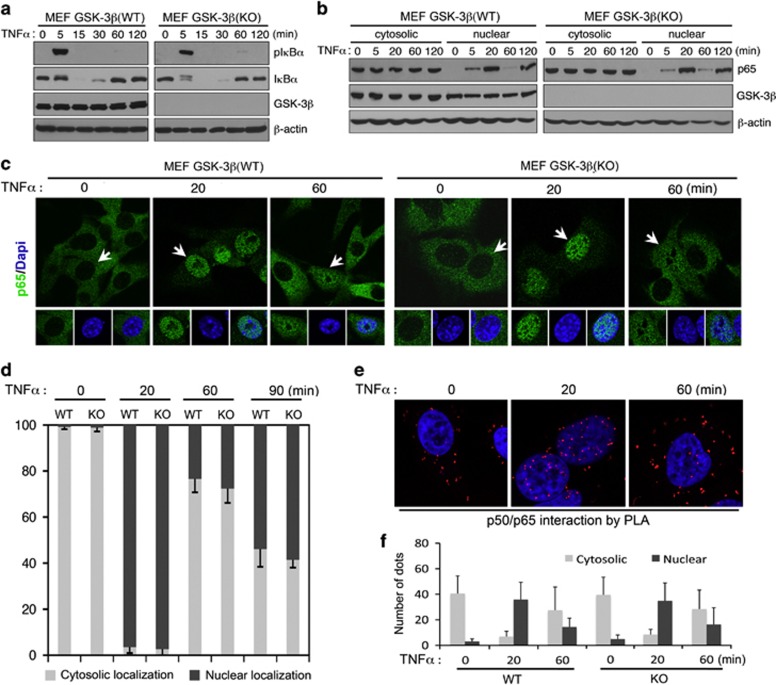
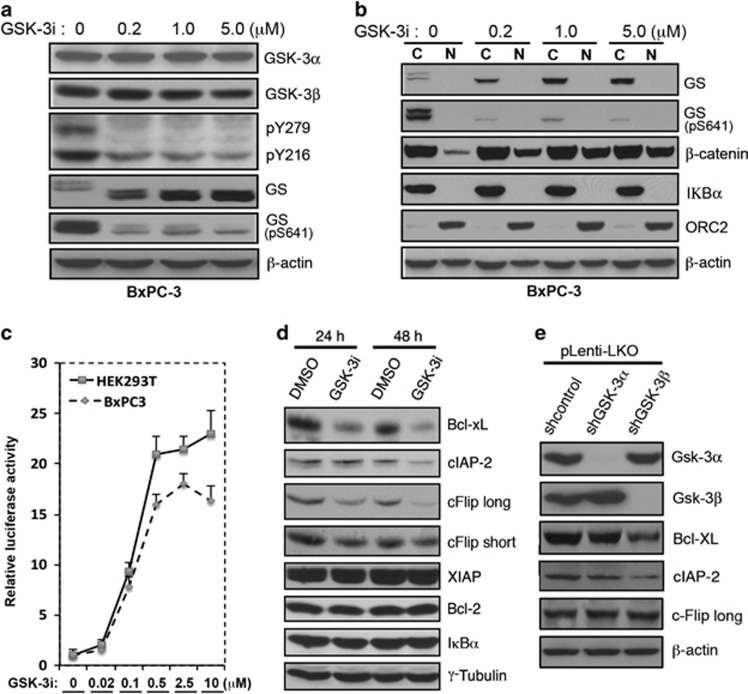
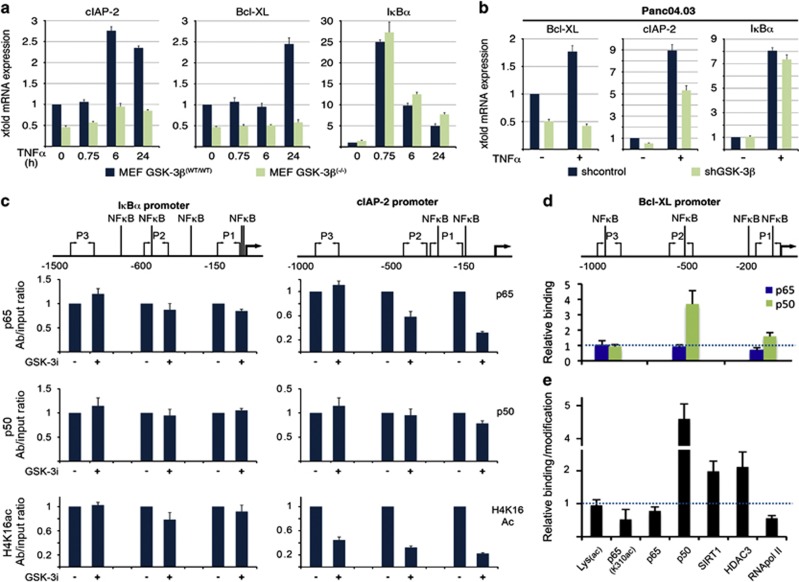
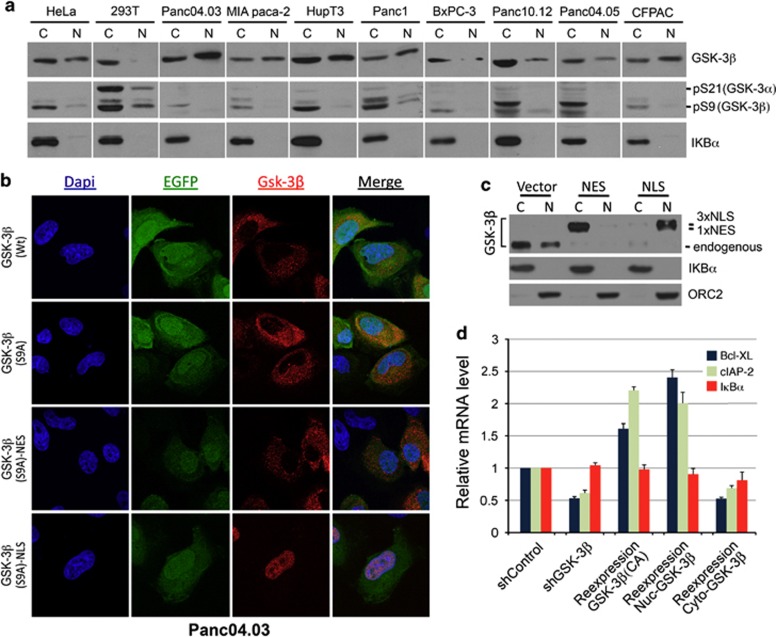
While TRAIL is a promising anticancer agent due to its ability to selectively induce apoptosis in neoplastic cells, many tumors, including pancreatic ductal adenocarcinoma (PDA), display intrinsic resistance, highlighting the need for TRAIL-sensitizing agents. Here we report that TRAIL-induced apoptosis in PDA cell lines is enhanced by pharmacological inhibition of glycogen synthase kinase-3 (GSK-3) or by shRNA-mediated depletion of either GSK-3α or GSK-3β. In contrast, depletion of GSK-3β, but not GSK-3α, sensitized PDA cell lines to TNFα-induced cell death. Further experiments demonstrated that TNFα-stimulated IκBα phosphorylation and degradation as well as p65 nuclear translocation were normal in GSK-3β-deficient MEFs. Nonetheless, inhibition of GSK-3β function in MEFs or PDA cell lines impaired the expression of the NF-κB target genes Bcl-xL and cIAP2, but not IκBα. Significantly, the expression of Bcl-xL and cIAP2 could be reestablished by expression of GSK-3β targeted to the nucleus but not GSK-3β targeted to the cytoplasm, suggesting that GSK-3β regulates NF-κB function within the nucleus. Consistent with this notion, chromatin immunoprecipitation demonstrated that GSK-3 inhibition resulted in either decreased p65 binding to the promoter of BIR3, which encodes cIAP2, or increased p50 binding as well as recruitment of SIRT1 and HDAC3 to the promoter of BCL2L1, which encodes Bcl-xL. Importantly, depletion of Bcl-xL but not cIAP2, mimicked the sensitizing effect of GSK-3 inhibition on TRAIL-induced apoptosis, whereas Bcl-xL overexpression ameliorated the sensitization by GSK-3 inhibition. These results not only suggest that GSK-3β overexpression and nuclear localization contribute to TNFα and TRAIL resistance via anti-apoptotic NF-κB genes such as Bcl-xL, but also provide a rationale for further exploration of GSK-3 inhibitors combined with TRAIL for the treatment of PDA.
虽然肿瘤坏死因子相关凋亡诱导配体(TRAIL)因其能够选择性地诱导肿瘤细胞凋亡而成为一种有前景的抗癌药物,但许多肿瘤,包括胰腺导管腺癌(PDA),都表现出内在抗性,这突出了对TRAIL增敏剂的需求。在此我们报告,通过糖原合酶激酶-3(GSK-3)的药理学抑制或通过shRNA介导的GSK-3α或GSK-3β的缺失,可增强TRAIL诱导的PDA细胞系凋亡。相反,GSK-3β的缺失而非GSK-3α的缺失使PDA细胞系对肿瘤坏死因子α(TNFα)诱导的细胞死亡敏感。进一步的实验表明,在GSK-3β缺陷的小鼠胚胎成纤维细胞(MEF)中,TNFα刺激的IκBα磷酸化和降解以及p65核转位是正常的。尽管如此,抑制MEF或PDA细胞系中的GSK-3β功能会损害核因子κB(NF-κB)靶基因Bcl-xL和细胞凋亡抑制蛋白2(cIAP2)的表达,但不会影响IκBα的表达。重要的是,通过靶向细胞核的GSK-3β的表达而非靶向细胞质的GSK-3β的表达可以重建Bcl-xL和cIAP2的表达,这表明GSK-3β在细胞核内调节NF-κB功能。与此观点一致,染色质免疫沉淀表明,GSK-3抑制导致p65与编码cIAP2的杆状病毒IAP重复序列3(BIR3)启动子的结合减少,或者导致p50结合增加以及沉默信息调节因子2相关酶1(SIRT1)和组蛋白去乙酰化酶3(HDAC3)募集到编码Bcl-xL的BCL2L1启动子上。重要的是,Bcl-xL而非cIAP2的缺失模拟了GSK-3抑制对TRAIL诱导凋亡的增敏作用,而Bcl-xL的过表达改善了GSK-3抑制的增敏作用。这些结果不仅表明GSK-3β的过表达和核定位通过抗凋亡的NF-κB基因如Bcl-xL导致对TNFα和TRAIL的抗性,而且为进一步探索GSK-3抑制剂与TRAIL联合用于治疗PDA提供了理论依据。