Berger Emily, Yorukoglu Deniz, Peng Jian, Berger Bonnie
Department of Mathematics, MIT, Cambridge, Massachusetts, United States of America; Computer Science and Artificial Intelligence Laboratory, MIT, Cambridge, Massachusetts, United States of America; Department of Mathematics, UC Berkeley, Berkeley, California, United States of America.
Computer Science and Artificial Intelligence Laboratory, MIT, Cambridge, Massachusetts, United States of America.
PLoS Comput Biol. 2014 Mar 27;10(3):e1003502. doi: 10.1371/journal.pcbi.1003502. eCollection 2014 Mar.
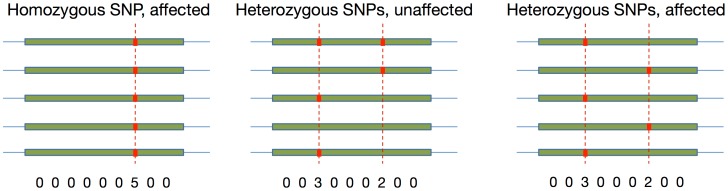
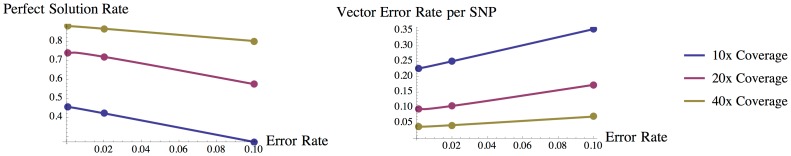
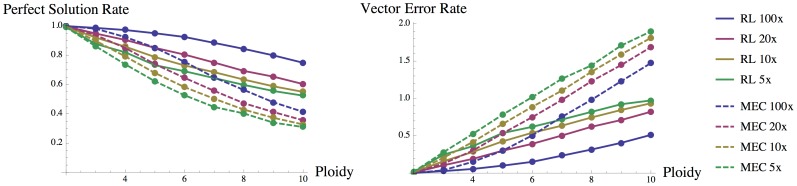
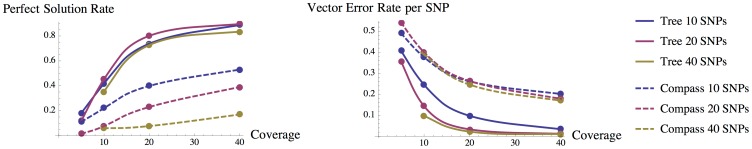
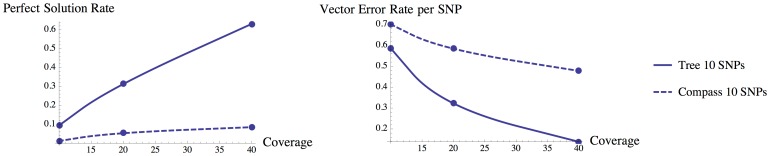
As the more recent next-generation sequencing (NGS) technologies provide longer read sequences, the use of sequencing datasets for complete haplotype phasing is fast becoming a reality, allowing haplotype reconstruction of a single sequenced genome. Nearly all previous haplotype reconstruction studies have focused on diploid genomes and are rarely scalable to genomes with higher ploidy. Yet computational investigations into polyploid genomes carry great importance, impacting plant, yeast and fish genomics, as well as the studies of the evolution of modern-day eukaryotes and (epi)genetic interactions between copies of genes. In this paper, we describe a novel maximum-likelihood estimation framework, HapTree, for polyploid haplotype assembly of an individual genome using NGS read datasets. We evaluate the performance of HapTree on simulated polyploid sequencing read data modeled after Illumina sequencing technologies. For triploid and higher ploidy genomes, we demonstrate that HapTree substantially improves haplotype assembly accuracy and efficiency over the state-of-the-art; moreover, HapTree is the first scalable polyplotyping method for higher ploidy. As a proof of concept, we also test our method on real sequencing data from NA12878 (1000 Genomes Project) and evaluate the quality of assembled haplotypes with respect to trio-based diplotype annotation as the ground truth. The results indicate that HapTree significantly improves the switch accuracy within phased haplotype blocks as compared to existing haplotype assembly methods, while producing comparable minimum error correction (MEC) values. A summary of this paper appears in the proceedings of the RECOMB 2014 conference, April 2-5.
随着更新的下一代测序(NGS)技术能够提供更长的读段序列,利用测序数据集进行完整单倍型定相正迅速成为现实,这使得对单个测序基因组进行单倍型重建成为可能。几乎所有之前的单倍型重建研究都集中在二倍体基因组上,很少能扩展到更高倍性的基因组。然而,对多倍体基因组的计算研究具有重要意义,它影响着植物、酵母和鱼类基因组学,以及现代真核生物进化和基因拷贝之间的(表观)遗传相互作用的研究。在本文中,我们描述了一种新颖的最大似然估计框架HapTree,用于使用NGS读段数据集对单个基因组进行多倍体单倍型组装。我们在模拟的、以Illumina测序技术为模型的多倍体测序读段数据上评估了HapTree的性能。对于三倍体及更高倍性的基因组,我们证明HapTree在单倍型组装准确性和效率方面比现有技术有显著提高;此外,HapTree是第一种可扩展的用于更高倍性的多倍体分型方法。作为概念验证,我们还在来自NA12878(千人基因组计划)的真实测序数据上测试了我们的方法,并以基于三联体的双倍型注释作为基准来评估组装单倍型的质量。结果表明,与现有的单倍型组装方法相比,HapTree显著提高了定相单倍型块内的切换准确性,同时产生了相当的最小错误校正(MEC)值。本文的摘要发表在2014年4月2 - 5日的RECOMB会议论文集上。