Al-Shibli Amar, Yusuf Madinah, Abounajab Issam, Willems Patrick J
GENDIA (GENetic DIAgnostic Network), Antwerp, Belgium.
Department of Academic Affairs, Tawam Hospital, Al-Ain, United Arab Emirates.
Springerplus. 2014 Feb 18;3:96. doi: 10.1186/2193-1801-3-96. eCollection 2014.
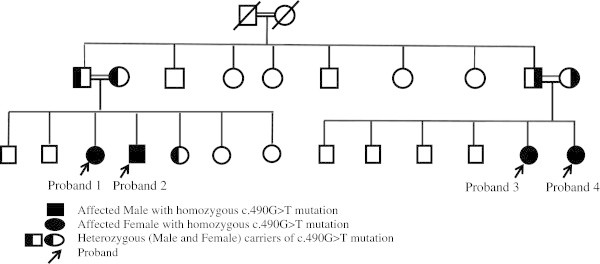
Patients with renal diseases associated with salt-losing tubulopathies categorized as Gitelman and classic form of Bartter syndrome have undergone genetic screening for possible mutation capture in two different genes: SLC12A3 and CLCNKB. Clinical symptoms of these two diseases may overlap. Bartter syndrome and Gitelman syndrome are autosomal recessive salt-losing tubulopathies with hypokalemia, metabolic alkalosis, hyperreninemia, hyperplasia of the juxtaglomerular apparatus, hyperaldosteronism, and, in some patients, hypomagnesemia. Here we describe four patients from an inbred family with a novel missense variant in the CLCNKB gene. All of patients are asymptomatic; yet they have the typical metabolic abnormality of salt losing tubulopathies. One of those patients had hypomagnesaemia while others not. Clinical and laboratory data of all patients was described. All 4 patients have a homozygous c.490G > T missense variant in exon 5 of the CLCNKB gene. This variant alters a glycine into a cysteine on amino acid position 164 of the resulting protein (p.Gly164Cys). The c.490G > T variant is a novel variant not previously described in other patients nor controls. Polyphen analysis predicts the variation to be possibly damaging. Analysis of SLC12A3 was normal. Here in we are describing a novel homozygous c.490G > T missense variation was identified in exon 5 of the CLCNKB gene was identified in an Emirati patients with a mild manifestation of Bartter - Gitelman syndrome.
患有与失盐性肾小管病相关的肾脏疾病(分类为吉特曼综合征和经典型巴特综合征)的患者,已针对两个不同基因(SLC12A3和CLCNKB)进行了可能的突变捕获基因筛查。这两种疾病的临床症状可能重叠。巴特综合征和吉特曼综合征是常染色体隐性失盐性肾小管病,伴有低钾血症、代谢性碱中毒、高肾素血症、肾小球旁器增生、醛固酮增多症,部分患者还伴有低镁血症。在此,我们描述了一个近亲家族中的四名患者,他们的CLCNKB基因存在一种新的错义变异。所有患者均无症状,但具有失盐性肾小管病的典型代谢异常。其中一名患者有低镁血症,其他患者没有。描述了所有患者的临床和实验室数据。所有4名患者在CLCNKB基因第5外显子中均有纯合的c.490G>T错义变异。该变异导致所产生蛋白质的第164位氨基酸上的甘氨酸变为半胱氨酸(p.Gly164Cys)。c.490G>T变异是一种新变异,此前在其他患者或对照中均未描述过。Polyphen分析预测该变异可能具有损害性。SLC12A3分析正常。在此,我们描述了在一名患有轻度巴特 - 吉特曼综合征表现的阿联酋患者中,在CLCNKB基因第5外显子中鉴定出一种新的纯合c.490G>T错义变异。