Sofou Kalliopi, De Coo Irenaeus F M, Isohanni Pirjo, Ostergaard Elsebet, Naess Karin, De Meirleir Linda, Tzoulis Charalampos, Uusimaa Johanna, De Angst Isabell B, Lönnqvist Tuula, Pihko Helena, Mankinen Katariina, Bindoff Laurence A, Tulinius Már, Darin Niklas
Department of Paediatrics, University of Gothenburg, The Queen Silvia's Children Hospital, SE-416 85 Gothenburg, Sweden.
Orphanet J Rare Dis. 2014 Apr 15;9:52. doi: 10.1186/1750-1172-9-52.
Leigh syndrome is a progressive neurodegenerative disorder, associated with primary or secondary dysfunction of the mitochondrial oxidative phosphorylation. Despite the fact that Leigh syndrome is the most common phenotype of mitochondrial disorders in children, longitudinal natural history data is missing. This study was undertaken to assess the phenotypic and genotypic spectrum of patients with Leigh syndrome, characterise the clinical course and identify predictors of survival in a large cohort of patients.
This is a retrospective study of patients with Leigh syndrome that have been followed at eight centers specialising in mitochondrial diseases in Europe; Gothenburg, Rotterdam, Helsinki, Copenhagen, Stockholm, Brussels, Bergen and Oulu.
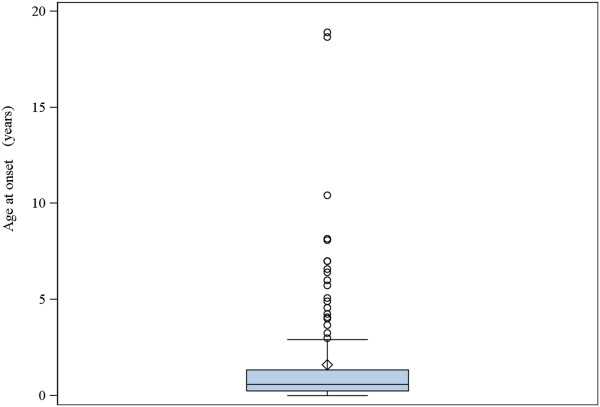
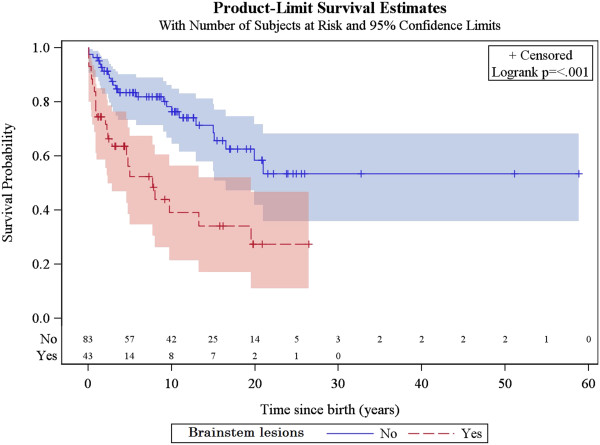
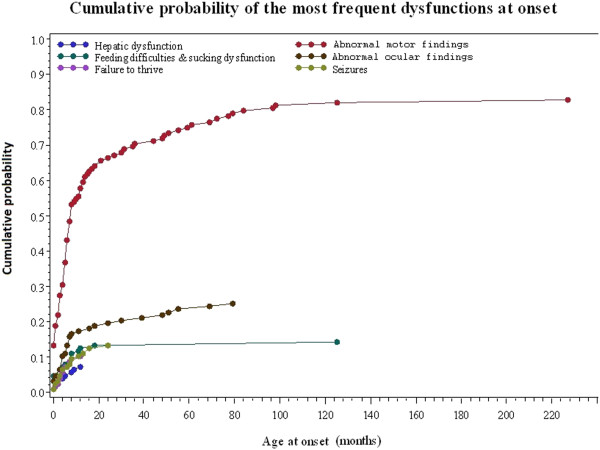
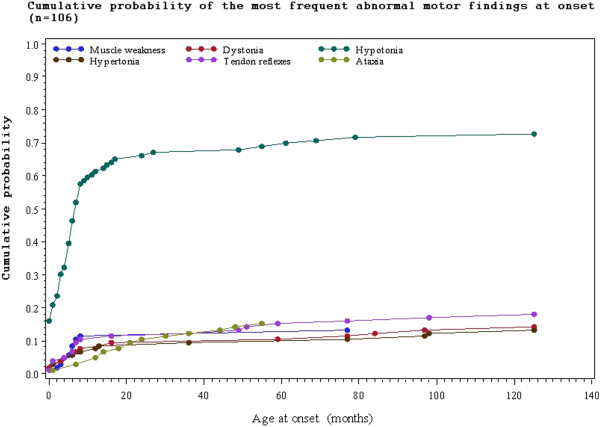
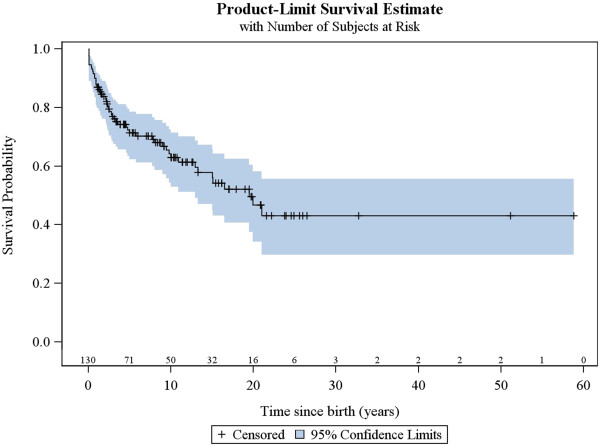
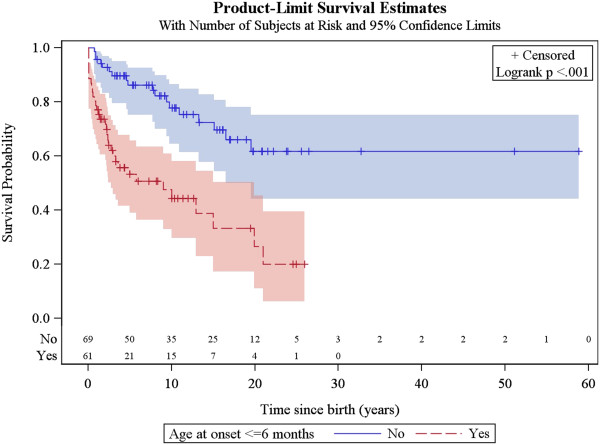
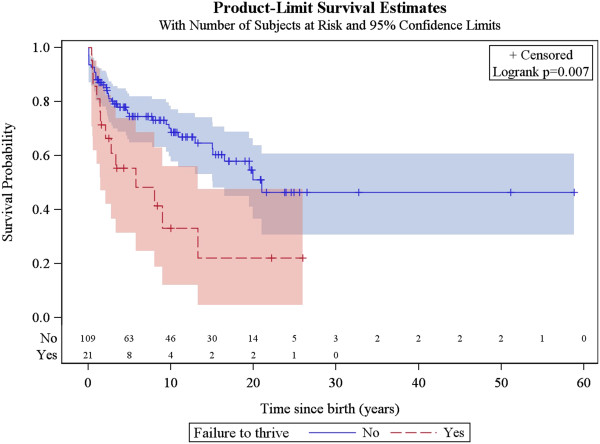
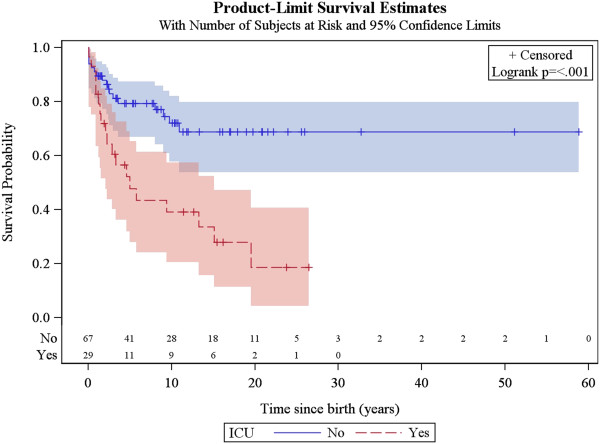
A total of 130 patients were included (78 males; 52 females), of whom 77 patients had identified pathogenic mutations. The median age of disease onset was 7 months, with 80.8% of patients presenting by the age of 2 years. The most common clinical features were abnormal motor findings, followed by abnormal ocular findings. Epileptic seizures were reported in 40% of patients. Approximately 44% of patients experienced acute exacerbations requiring hospitalisation during the previous year, mainly due to infections. The presence of pathological signs at birth and a history of epileptic seizures were associated with higher occurrence of acute exacerbations and/or relapses. Increased lactate in the cerebrospinal fluid was significantly correlated to a more severe disease course, characterised by early onset before 6 months of age, acute exacerbations and/or relapses, as well as brainstem involvement. 39% of patients had died by the age of 21 years, at a median age of 2.4 years. Disease onset before 6 months of age, failure to thrive, brainstem lesions on neuroimaging and intensive care treatment were significantly associated with poorer survival.
This is a multicenter study performed in a large cohort of patients with Leigh syndrome. Our data help define the natural history of Leigh syndrome and identify novel predictors of disease severity and long-term prognosis.
Leigh综合征是一种进行性神经退行性疾病,与线粒体氧化磷酸化的原发性或继发性功能障碍相关。尽管Leigh综合征是儿童线粒体疾病最常见的表型,但缺乏纵向自然史数据。本研究旨在评估Leigh综合征患者的表型和基因型谱,描述临床病程,并确定一大群患者的生存预测因素。
这是一项对在欧洲八个线粒体疾病专科中心(哥德堡、鹿特丹、赫尔辛基、哥本哈根、斯德哥尔摩、布鲁塞尔、卑尔根和奥卢)随访的Leigh综合征患者进行的回顾性研究。
共纳入130例患者(78例男性;52例女性),其中77例患者已鉴定出致病突变。疾病发病的中位年龄为7个月,80.8%的患者在2岁前发病。最常见的临床特征是运动异常,其次是眼部异常。40%的患者报告有癫痫发作。约44%的患者在前一年经历了需要住院治疗的急性加重,主要原因是感染。出生时存在病理体征和癫痫发作史与急性加重和/或复发的发生率较高相关。脑脊液中乳酸水平升高与更严重的病程显著相关,其特征为6个月前发病早、急性加重和/或复发以及脑干受累。39%的患者在21岁前死亡,中位年龄为2.4岁。6个月前发病、发育不良、神经影像学显示脑干病变和重症监护治疗与较差的生存率显著相关。
这是一项在一大群Leigh综合征患者中进行的多中心研究。我们的数据有助于定义Leigh综合征的自然史,并确定疾病严重程度和长期预后的新预测因素。