Ohmiya Hiroko, Vitezic Morana, Frith Martin C, Itoh Masayoshi, Carninci Piero, Forrest Alistair R R, Hayashizaki Yoshihide, Lassmann Timo
RIKEN Center for Life Science Technologies (CLST), Division of Genomic Technologies, RIKEN Yokohama Institute, 1-7-22 Suehiro-cho, Tsurumi-ku, 230-0045 Yokohama, Japan.
BMC Genomics. 2014 Apr 25;15:269. doi: 10.1186/1471-2164-15-269.
Next generation sequencing based technologies are being extensively used to study transcriptomes. Among these, cap analysis of gene expression (CAGE) is specialized in detecting the most 5' ends of RNA molecules. After mapping the sequenced reads back to a reference genome CAGE data highlights the transcriptional start sites (TSSs) and their usage at a single nucleotide resolution.
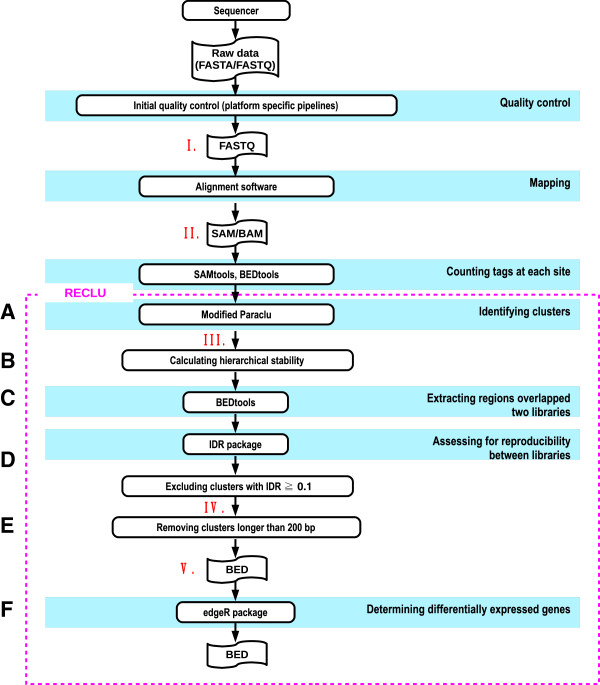
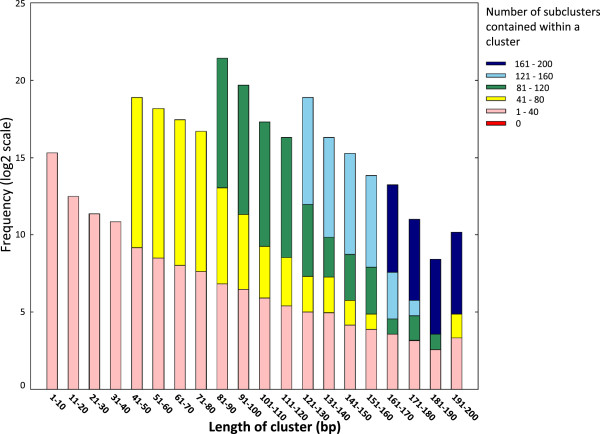
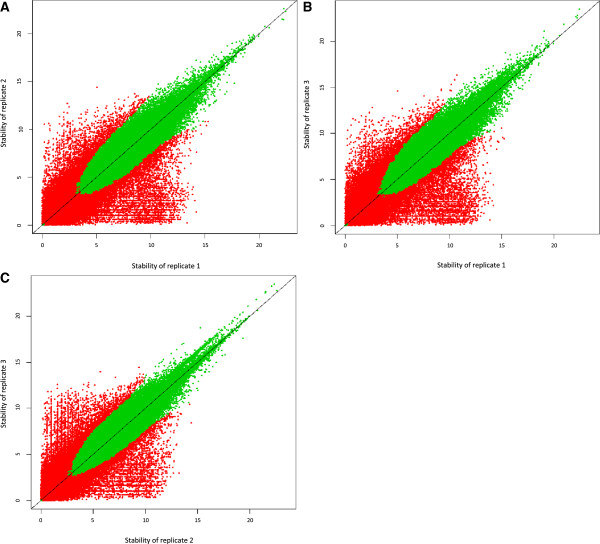
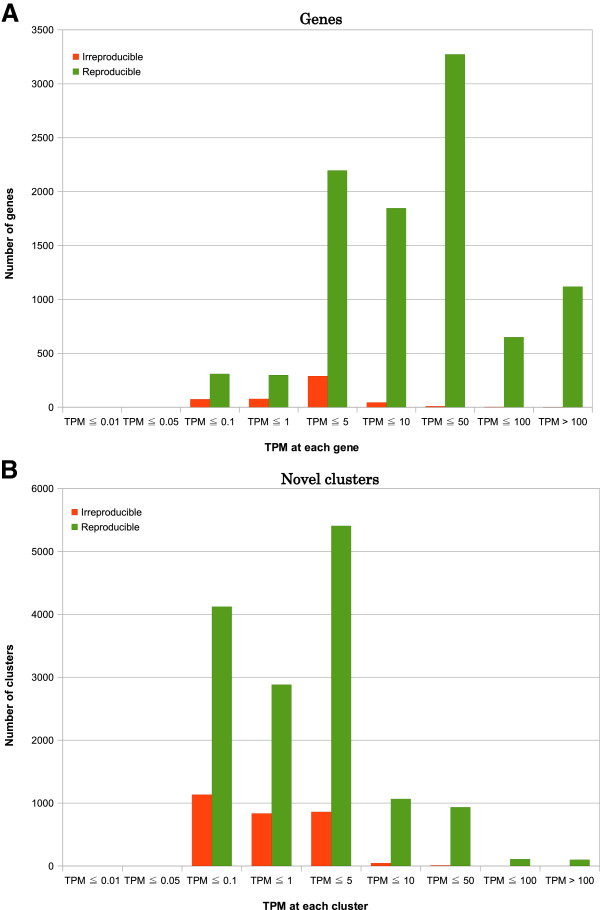
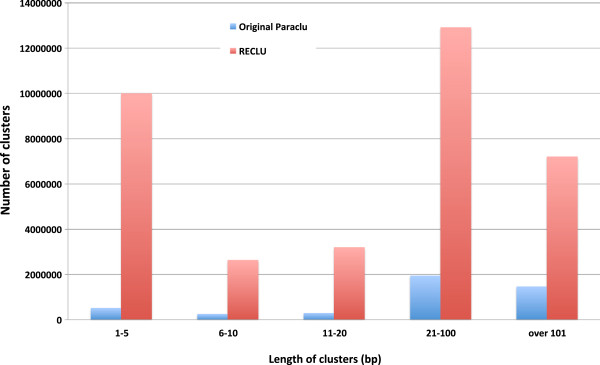
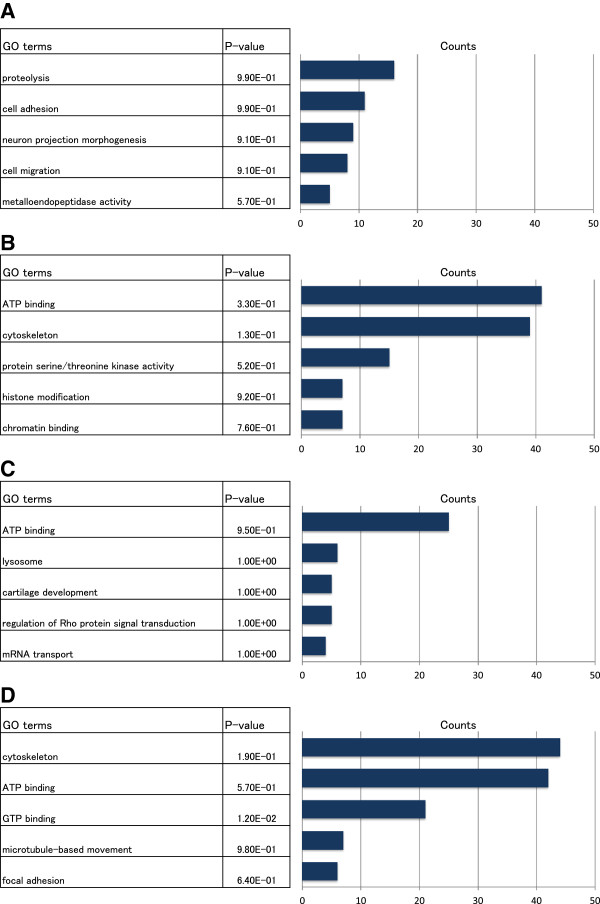
We propose a pipeline to group the single nucleotide TSS into larger reproducible peaks and compare their usage across biological states. Importantly, our pipeline discovers broad peaks as well as the fine structure of individual transcriptional start sites embedded within them. We assess the performance of our approach on a large CAGE datasets including 156 primary cell types and two cell lines with biological replicas. We demonstrate that genes have complicated structures of transcription initiation events. In particular, we discover that narrow peaks embedded in broader regions of transcriptional activity can be differentially used even if the larger region is not.
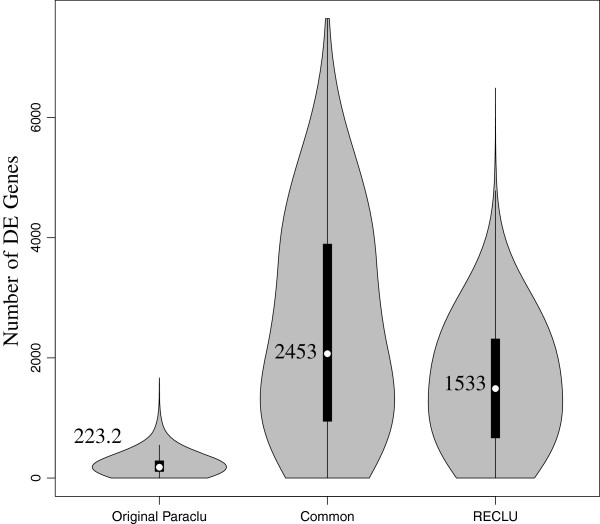
By examining the reproducible fine scaled organization of TSS we can detect many differentially regulated peaks undetected by previous approaches.
基于新一代测序的技术正被广泛用于研究转录组。其中,基因表达的帽分析(CAGE)专门用于检测RNA分子的最5'端。将测序读段映射回参考基因组后,CAGE数据以单核苷酸分辨率突出显示转录起始位点(TSS)及其使用情况。
我们提出了一种流程,将单核苷酸TSS分组为更大的可重复峰,并比较它们在不同生物学状态下的使用情况。重要的是,我们的流程发现了宽峰以及嵌入其中的单个转录起始位点的精细结构。我们在包括156种原代细胞类型和两个带有生物学重复的细胞系的大型CAGE数据集上评估了我们方法的性能。我们证明基因具有复杂的转录起始事件结构。特别是,我们发现即使较大区域没有差异使用,嵌入转录活性较宽区域的窄峰也可能被差异使用。
通过检查TSS的可重复精细尺度组织,我们可以检测到许多以前的方法未检测到的差异调节峰。