Aguirre-Soto Alan, Lim Chern-Hooi, Hwang Albert T, Musgrave Charles B, Stansbury Jeffrey W
Department of Chemical and Biological Engineering, University of Colorado Boulder , 3415 Colorado Ave., Boulder, Colorado 80303, United States.
J Am Chem Soc. 2014 May 21;136(20):7418-27. doi: 10.1021/ja502441d. Epub 2014 May 8.
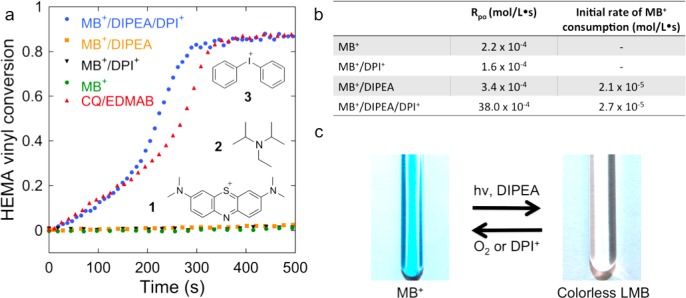
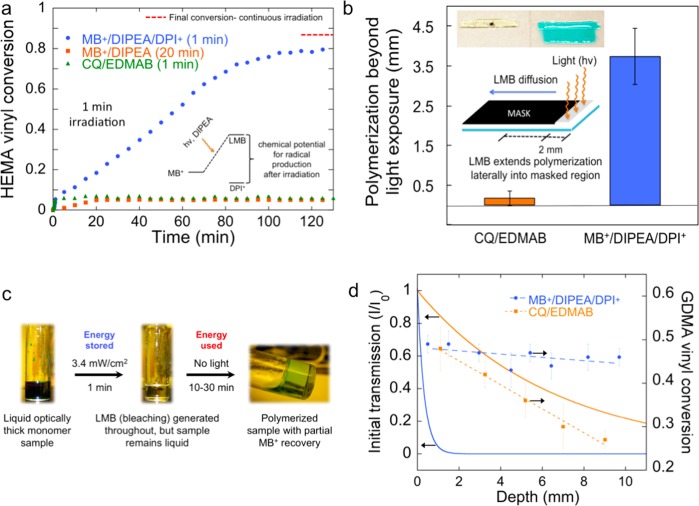
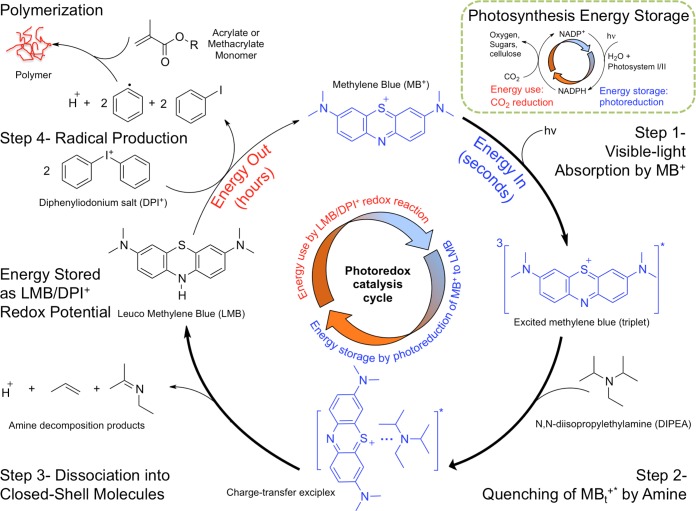
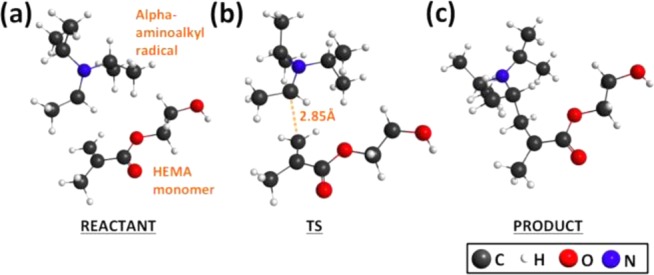
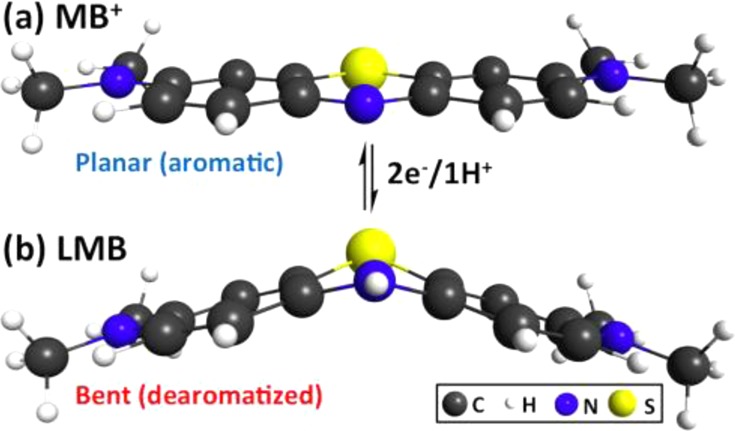
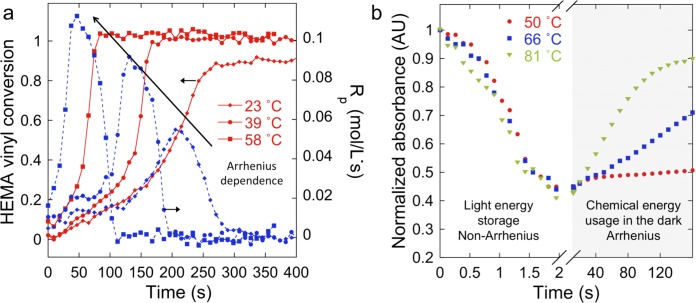
We report the latent production of free radicals from energy stored in a redox potential through a 2e(-)/1H(+) transfer process, analogous to energy harvesting in photosynthesis, using visible-light organic photoredox catalysis (photocatalysis) of methylene blue chromophore with a sacrificial sterically hindered amine reductant and an onium salt oxidant. This enables light-initiated free-radical polymerization to continue over extended time intervals (hours) in the dark after brief (seconds) low-intensity illumination and beyond the spatial reach of light by diffusion of the metastable leuco-methylene blue photoproduct. The present organic photoredox catalysis system functions via a 2e(-)/1H(+) shuttle mechanism, as opposed to the 1e(-) transfer process typical of organometallic-based and conventional organic multicomponent photoinitiator formulations. This prevents immediate formation of open-shell (radical) intermediates from the amine upon light absorption and enables the "storage" of light-energy without spontaneous initiation of the polymerization. Latent energy release and radical production are then controlled by the subsequent light-independent reaction (analogous to the Calvin cycle) between leuco-methylene blue and the onium salt oxidant that is responsible for regeneration of the organic methylene blue photocatalyst. This robust approach for photocatalysis-based energy harvesting and extended release in the dark enables temporally controlled redox initiation of polymer syntheses under low-intensity short exposure conditions and permits visible-light-mediated synthesis of polymers at least 1 order of magnitude thicker than achievable with conventional photoinitiated formulations and irradiation regimes.
我们报道了通过2e(-)/1H(+)转移过程,从氧化还原电位中储存的能量中潜在地产生自由基,这类似于光合作用中的能量收集,使用亚甲基蓝发色团的可见光有机光氧化还原催化(光催化),牺牲性空间位阻胺还原剂和鎓盐氧化剂。这使得光引发的自由基聚合在短暂(秒)的低强度光照后,能在黑暗中持续较长时间间隔(小时),并通过亚稳无色亚甲基蓝光产物的扩散,超越光的空间范围。目前的有机光氧化还原催化体系通过2e(-)/1H(+)穿梭机制起作用,这与基于有机金属和传统有机多组分光引发剂配方典型的1e(-)转移过程相反。这防止了胺在光吸收后立即形成开壳(自由基)中间体,并使得光能能够“储存”而不会自发引发聚合反应。潜在的能量释放和自由基产生随后由无色亚甲基蓝与负责再生有机亚甲基蓝光催化剂的鎓盐氧化剂之间随后的光独立反应(类似于卡尔文循环)控制。这种基于光催化的能量收集和黑暗中延长释放的稳健方法,能够在低强度短曝光条件下对聚合物合成进行时间控制的氧化还原引发,并允许可见光介导合成比传统光引发配方和辐照方式所能达到的至少厚1个数量级的聚合物。