West James, Austin Eric, Fessel Joshua P, Loyd James, Hamid Rizwan
Department of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Department of Pediatrics, Vanderbilt University Medical Center, Nashville, TN 37232, USA.
Drug Discov Today. 2014 Aug;19(8):1241-5. doi: 10.1016/j.drudis.2014.04.015. Epub 2014 May 2.
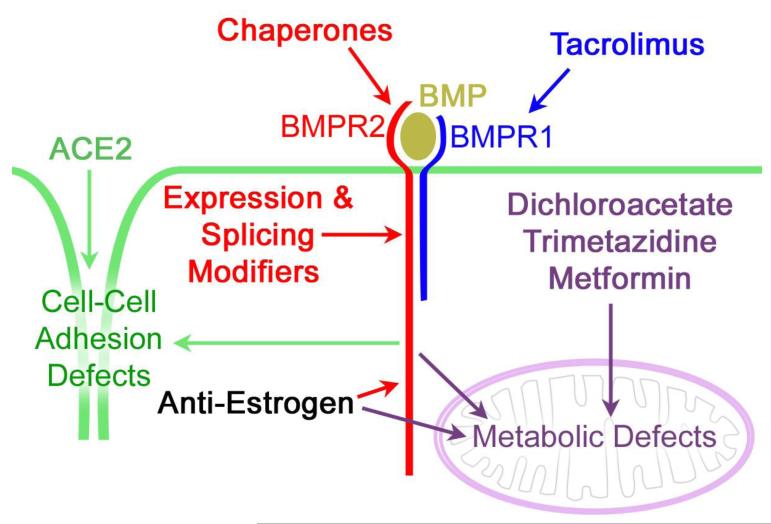
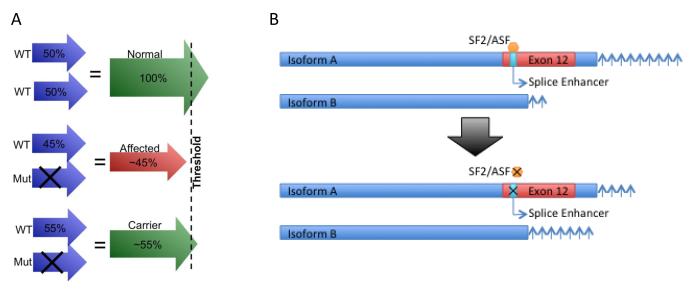
Pulmonary arterial hypertension (PAH) is a lethal disorder characterized by pulmonary arterial remodeling, increased right ventricular systolic pressure (RVSP), vasoconstriction and inflammation. The heritable form of PAH (HPAH) is usually (>80%) caused by mutations in the bone morphogenic protein receptor 2 (BMPR2) gene. Existing treatments for PAH typically focus on the end-stage sequelae of the disease, but do not address underlying mechanisms of vascular obstruction and blood flow and thus, in the long run, have limited effect because they treat the symptoms rather than the cause. Over the past decade, improved understanding of the molecular mechanisms behind the disease has enabled us to consider several novel therapeutic pathways. These include approaches directed toward BMPR2 gene expression, alternative splicing, downstream BMP signaling, metabolic pathways and the role of estrogens and estrogenic compounds in BMP signaling. It is likely that, ultimately, only one or two of these pathways will generate meaningful treatment options, however the potential benefits to PAH patients are still likely to be significant.
肺动脉高压(PAH)是一种致命性疾病,其特征为肺动脉重塑、右心室收缩压(RVSP)升高、血管收缩和炎症。遗传性PAH(HPAH)通常(>80%)由骨形态发生蛋白受体2(BMPR2)基因突变引起。PAH的现有治疗方法通常侧重于该疾病的终末期后遗症,但未涉及血管阻塞和血流的潜在机制,因此从长远来看,效果有限,因为它们只是治疗症状而非病因。在过去十年中,对该疾病背后分子机制的深入了解使我们能够考虑几种新的治疗途径。这些途径包括针对BMPR2基因表达、可变剪接、BMP下游信号传导、代谢途径以及雌激素和雌激素化合物在BMP信号传导中的作用的方法。最终,这些途径中可能只有一两种会产生有意义的治疗选择,然而对PAH患者的潜在益处可能仍然很大。