Department of Bioinformatics, Institute of Microbiology and Genetics, University of Göttingen, Goldschmidtstr. 1, 37073 Göttingen, Germany and Laboratoire Statistique et Génome, Université d'Évry Val d'Essonne, UMR CNRS 8071, USC INRA, 23 Boulevard de France, 91037 Évry, France.
Department of Bioinformatics, Institute of Microbiology and Genetics, University of Göttingen, Goldschmidtstr. 1, 37073 Göttingen, Germany and Laboratoire Statistique et Génome, Université d'Évry Val d'Essonne, UMR CNRS 8071, USC INRA, 23 Boulevard de France, 91037 Évry, FranceDepartment of Bioinformatics, Institute of Microbiology and Genetics, University of Göttingen, Goldschmidtstr. 1, 37073 Göttingen, Germany and Laboratoire Statistique et Génome, Université d'Évry Val d'Essonne, UMR CNRS 8071, USC INRA, 23 Boulevard de France, 91037 Évry, France.
Bioinformatics. 2014 Jul 15;30(14):2000-8. doi: 10.1093/bioinformatics/btu331. Epub 2014 May 13.
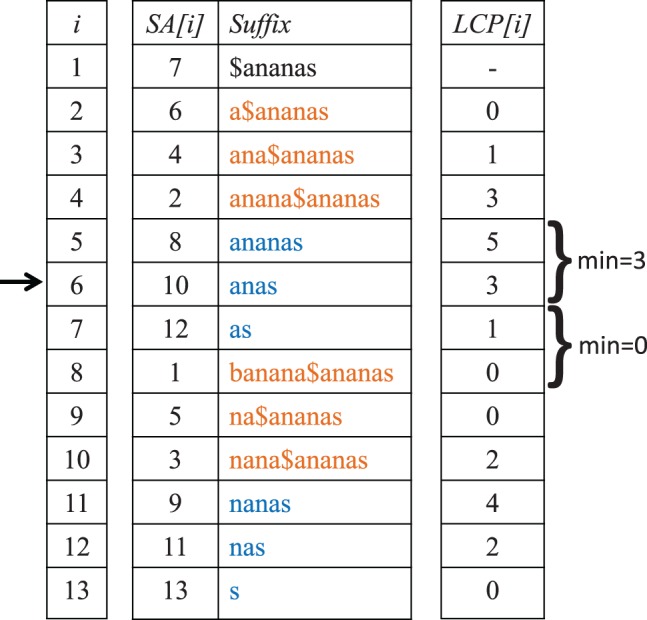
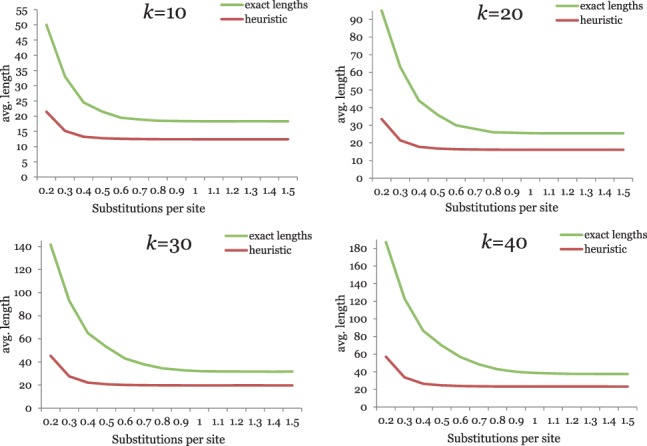
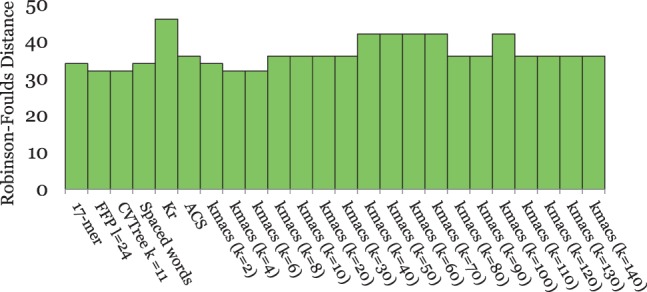
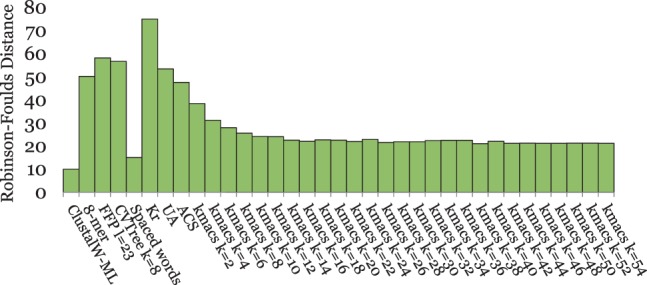
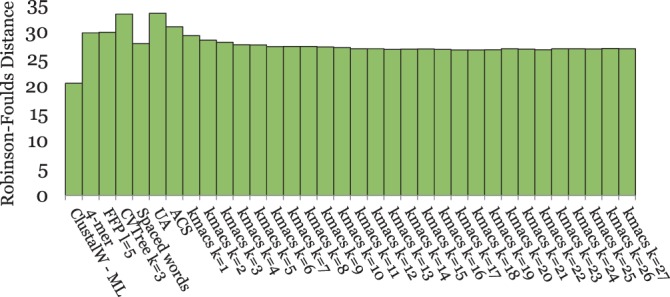
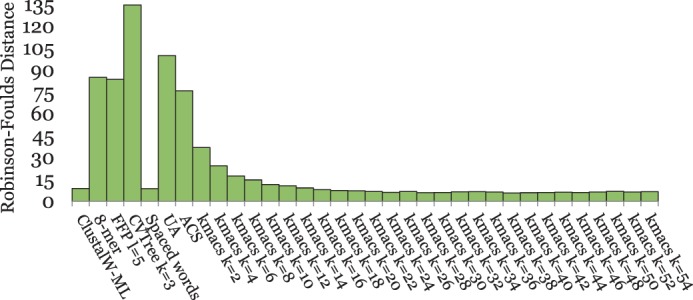
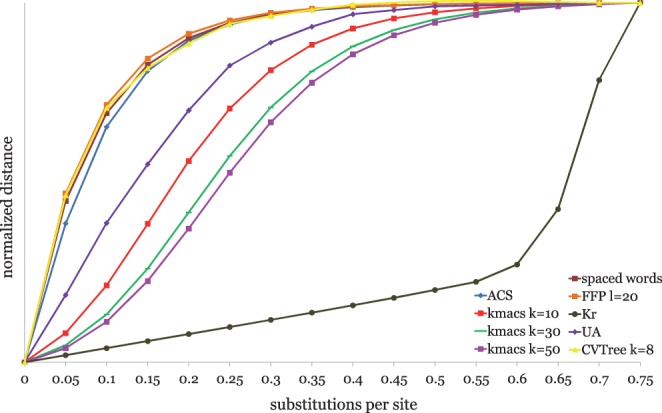
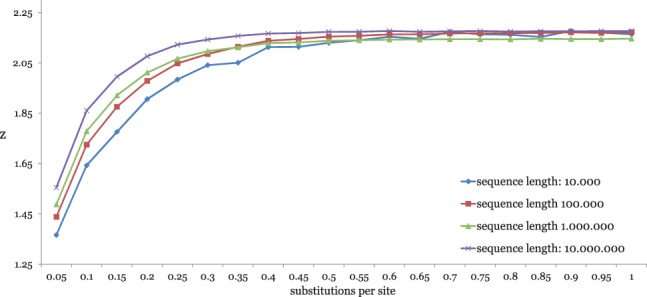
Alignment-based methods for sequence analysis have various limitations if large datasets are to be analysed. Therefore, alignment-free approaches have become popular in recent years. One of the best known alignment-free methods is the average common substring approach that defines a distance measure on sequences based on the average length of longest common words between them. Herein, we generalize this approach by considering longest common substrings with k mismatches. We present a greedy heuristic to approximate the length of such k-mismatch substrings, and we describe kmacs, an efficient implementation of this idea based on generalized enhanced suffix arrays.
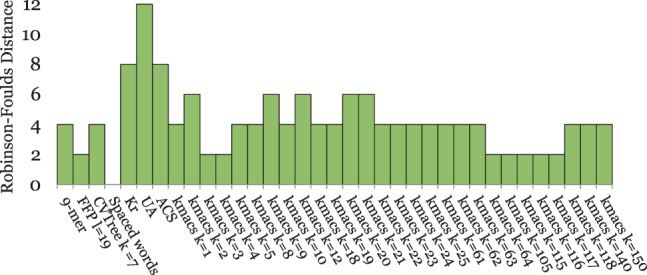
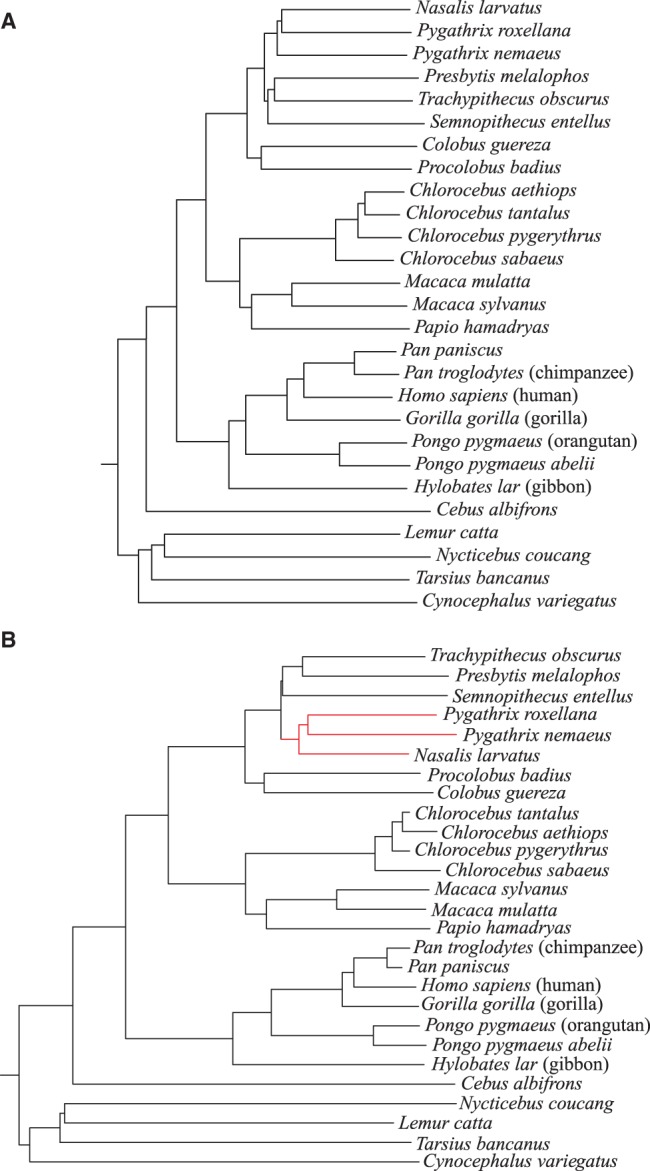
To evaluate the performance of our approach, we applied it to phylogeny reconstruction using a large number of DNA and protein sequence sets. In most cases, phylogenetic trees calculated with kmacs were more accurate than trees produced with established alignment-free methods that are based on exact word matches. Especially on protein sequences, our method seems to be superior. On simulated protein families, kmacs even outperformed a classical approach to phylogeny reconstruction using multiple alignment and maximum likelihood.
kmacs is implemented in C++, and the source code is freely available at http://kmacs.gobics.de/.
如果要分析大型数据集,基于比对的序列分析方法有各种局限性。因此,近年来无比对方法变得流行起来。其中最著名的无比对方法之一是平均公共子串方法,它基于它们之间最长公共单词的平均长度定义序列之间的距离度量。在此,我们通过考虑具有 k 个错配的最长公共子串来推广该方法。我们提出了一种贪婪启发式算法来近似这种 k-错配子串的长度,并描述了基于广义增强后缀数组的这种思想的有效实现 kmacs。
为了评估我们方法的性能,我们将其应用于使用大量 DNA 和蛋白质序列集进行系统发育重建。在大多数情况下,使用 kmacs 计算的系统发育树比基于精确单词匹配的基于比对的现有无比对方法生成的树更准确。特别是在蛋白质序列上,我们的方法似乎更优越。在模拟的蛋白质家族中,kmacs 甚至优于使用多重比对和最大似然进行系统发育重建的经典方法。
kmacs 是用 C++实现的,源代码可在 http://kmacs.gobics.de/ 免费获取。