University of Göttingen, Institute of Microbiology and Genetics, Department of Bioinformatics, Goldschmidtstraße 1, 37073 Göttingen, Germany
University of Göttingen, Institute of Microbiology and Genetics, Department of Bioinformatics, Goldschmidtstraße 1, 37073 Göttingen, Germany.
Nucleic Acids Res. 2014 Jul;42(Web Server issue):W7-11. doi: 10.1093/nar/gku398. Epub 2014 May 14.
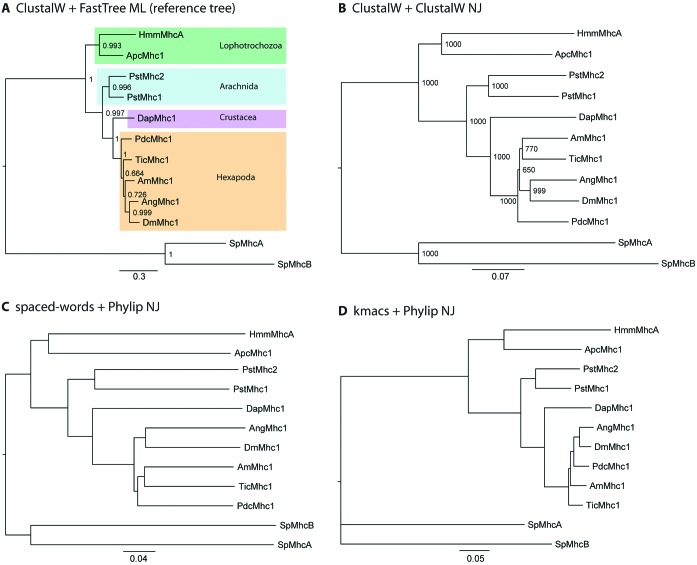
In this article, we present a user-friendly web interface for two alignment-free sequence-comparison methods that we recently developed. Most alignment-free methods rely on exact word matches to estimate pairwise similarities or distances between the input sequences. By contrast, our new algorithms are based on inexact word matches. The first of these approaches uses the relative frequencies of so-called spaced words in the input sequences, i.e. words containing 'don't care' or 'wildcard' symbols at certain pre-defined positions. Various distance measures can then be defined on sequences based on their different spaced-word composition. Our second approach defines the distance between two sequences by estimating for each position in the first sequence the length of the longest substring at this position that also occurs in the second sequence with up to k mismatches. Both approaches take a set of deoxyribonucleic acid (DNA) or protein sequences as input and return a matrix of pairwise distance values that can be used as a starting point for clustering algorithms or distance-based phylogeny reconstruction. The two alignment-free programmes are accessible through a web interface at 'Göttingen Bioinformatics Compute Server (GOBICS)': http://spaced.gobics.de http://kmacs.gobics.de and the source codes can be downloaded.
在本文中,我们为最近开发的两种无比对序列比较方法提供了一个用户友好的网络界面。大多数无比对方法依赖于精确的单词匹配来估计输入序列之间的两两相似性或距离。相比之下,我们的新算法基于不精确的单词匹配。第一种方法使用输入序列中所谓的间隔字的相对频率,即包含“不在乎”或“通配符”符号的字在某些预定义位置。然后可以基于它们不同的间隔字组成定义序列上的各种距离度量。我们的第二种方法通过估计在第一个序列中的每个位置,来定义两个序列之间的距离,即估计在该位置上的第一个序列中的最长子串的长度,该子串在第二个序列中也有最多 k 个不匹配。这两种无比对方法都以一组脱氧核糖核酸(DNA)或蛋白质序列作为输入,并返回一个两两距离值矩阵,可作为聚类算法或基于距离的系统发育重建的起点。这两个无比对程序可通过“哥廷根生物信息学计算服务器(GOBICS)”的网络界面访问:http://spaced.gobics.de/http://kmacs.gobics.de ,也可以下载源代码。