Department of Pathology, Stanford University Medical Center , Stanford, CA , USA.
Department of Pediatrics, Stanford University Medical Center , Stanford, CA , USA.
PeerJ. 2014 May 8;2:e384. doi: 10.7717/peerj.384. eCollection 2014.
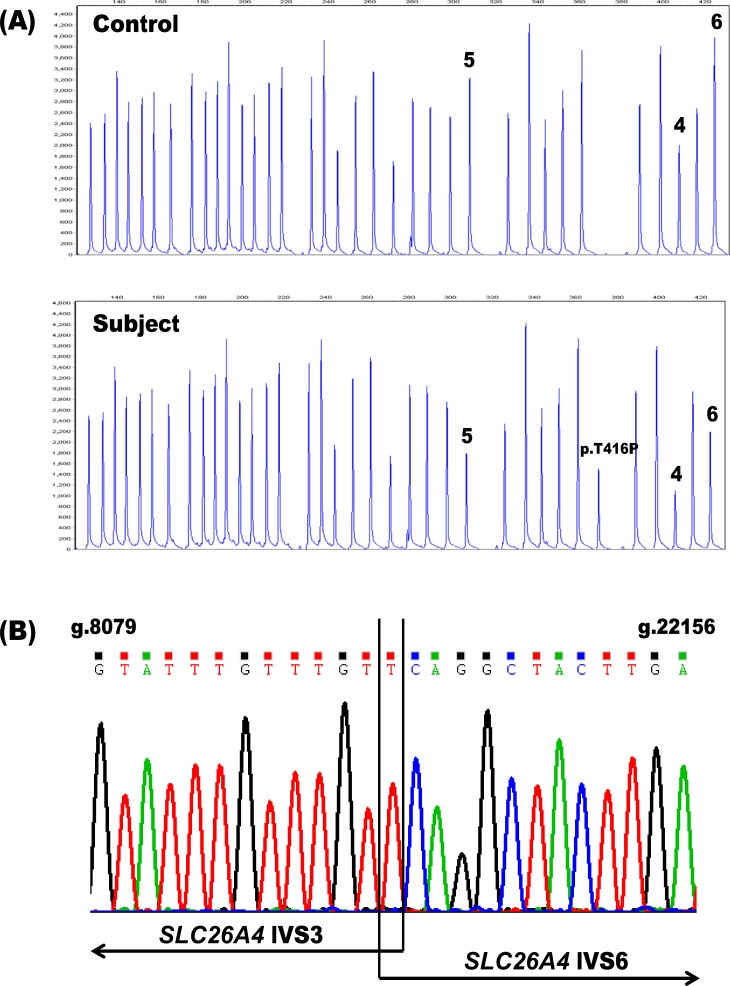
Pendred syndrome (PDS) and DFNB4 comprise a phenotypic spectrum of sensorineural hearing loss disorders that typically result from biallelic mutations of the SLC26A4 gene. Although PDS and DFNB4 are recessively inherited, sequencing of the coding regions and splice sites of SLC26A4 in individuals suspected to be affected with these conditions often fails to identify two mutations. We investigated the potential contribution of large SLC26A4 deletions and duplications to sensorineural hearing loss (SNHL) by screening 107 probands with one known SLC26A4 mutation by Multiplex Ligation-dependent Probe Amplification (MLPA). A heterozygous deletion, spanning exons 4-6, was detected in only one individual, accounting for approximately 1% of the missing mutations in our cohort. This low frequency is consistent with previously published MLPA results. We also examined the potential involvement of digenic inheritance in PDS/DFNB4 by sequencing the coding regions of FOXI1 and KCNJ10. Of the 29 probands who were sequenced, three carried nonsynonymous variants including one novel sequence change in FOXI1 and two polymorphisms in KCNJ10. We performed a review of prior studies and, in conjunction with our current data, conclude that the frequency of FOXI1 (1.4%) and KCNJ10 (3.6%) variants in PDS/DFNB4 individuals is low. Our results, in combination with previously published reports, indicate that large SLC26A4 deletions and duplications as well as mutations of FOXI1 and KCNJ10 play limited roles in the pathogenesis of SNHL and suggest that other genetic factors likely contribute to the phenotype.
皮特-霍恩综合征(PDS)和 DFNB4 构成了一种感觉神经性听力损失疾病的表型谱,其通常由 SLC26A4 基因的双等位基因突变引起。尽管 PDS 和 DFNB4 是隐性遗传的,但对疑似受这些病症影响的个体的 SLC26A4 编码区和剪接位点进行测序,往往无法发现两种突变。我们通过多重连接依赖性探针扩增(MLPA)筛查了 107 名携带一种已知 SLC26A4 突变的先证者,以研究大的 SLC26A4 缺失和重复是否会导致感觉神经性听力损失(SNHL)。仅在一名个体中检测到跨越外显子 4-6 的杂合性缺失,在我们的队列中,该缺失大约占缺失突变的 1%。这一低频率与之前发表的 MLPA 结果一致。我们还通过对 FOXI1 和 KCNJ10 的编码区进行测序,研究了 PDS/DFNB4 中双基因遗传的潜在参与。在被测序的 29 名先证者中,有 3 名携带非同义变异,包括 FOXI1 中的一个新序列变化和 KCNJ10 中的两个多态性。我们对之前的研究进行了回顾,并结合我们当前的数据得出结论,PDS/DFNB4 个体中 FOXI1(1.4%)和 KCNJ10(3.6%)变异的频率较低。我们的结果与之前发表的报告相结合,表明 SLC26A4 缺失和重复以及 FOXI1 和 KCNJ10 的突变在 SNHL 的发病机制中作用有限,并表明其他遗传因素可能对表型有贡献。