Department of Biomolecular Chemistry, University of Wisconsin, Madison, WI 53706, USA ; Wisconsin Institute for Discovery, University of Wisconsin, Madison, WI 53715, USA.
Wisconsin Institute for Discovery, University of Wisconsin, Madison, WI 53715, USA ; Biotechnology Center, University of Wisconsin, Madison, WI 53706, USA.
Epigenetics Chromatin. 2014 Apr 24;7:7. doi: 10.1186/1756-8935-7-7. eCollection 2014.
Histone post-translational modifications (PTMs) are key epigenetic regulators in chromatin-based processes. Increasing evidence suggests that vast combinations of PTMs exist within chromatin histones. These complex patterns, rather than individual PTMs, are thought to define functional chromatin states. However, the ability to interrogate combinatorial histone PTM patterns at the nucleosome level has been limited by the lack of direct molecular tools.
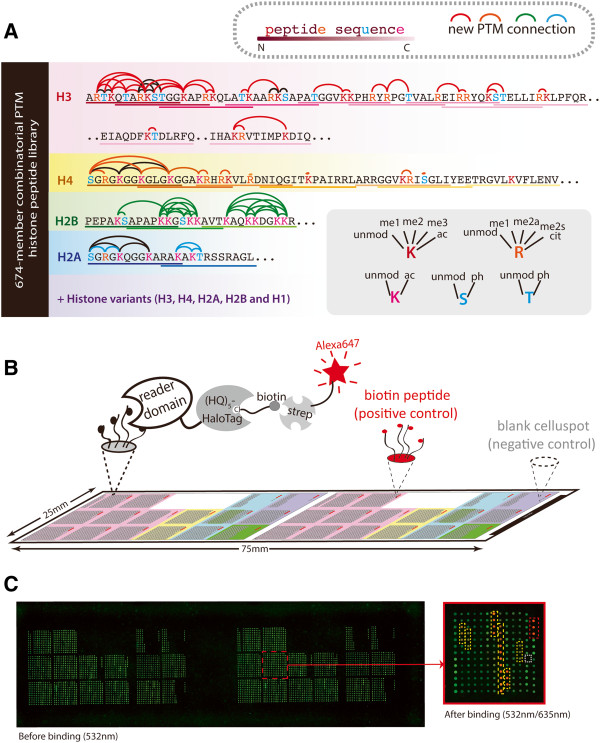
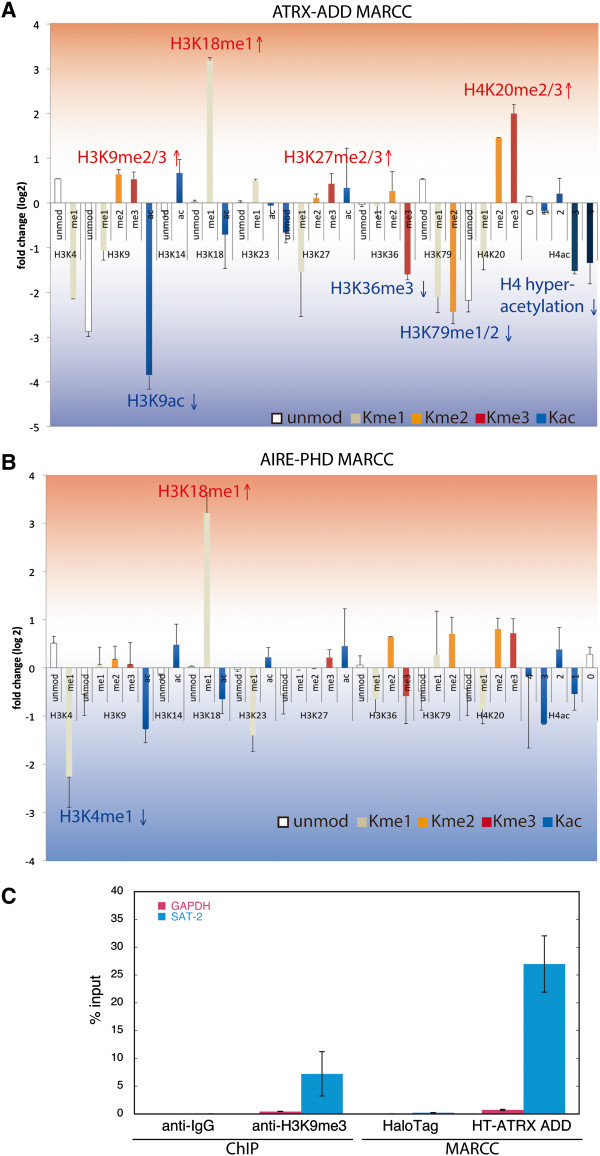
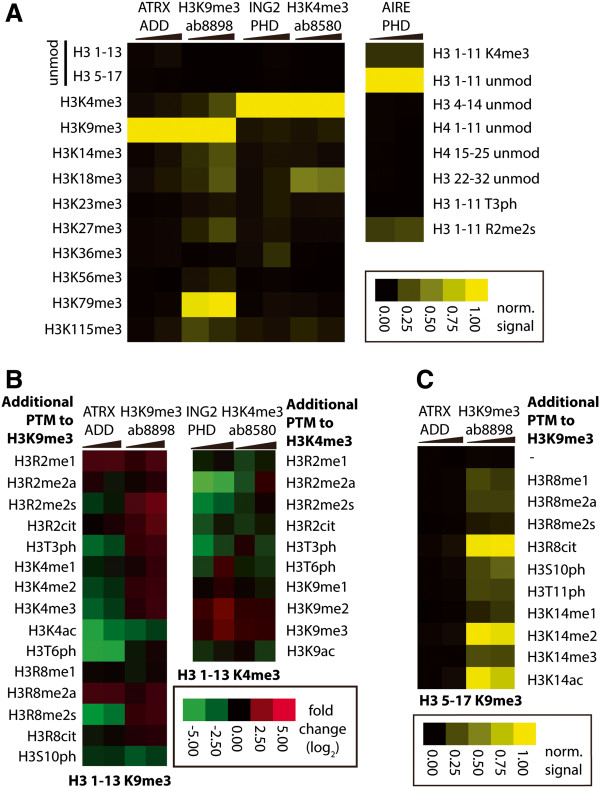
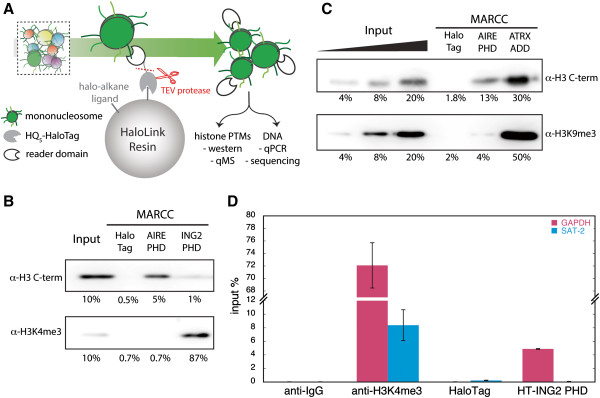
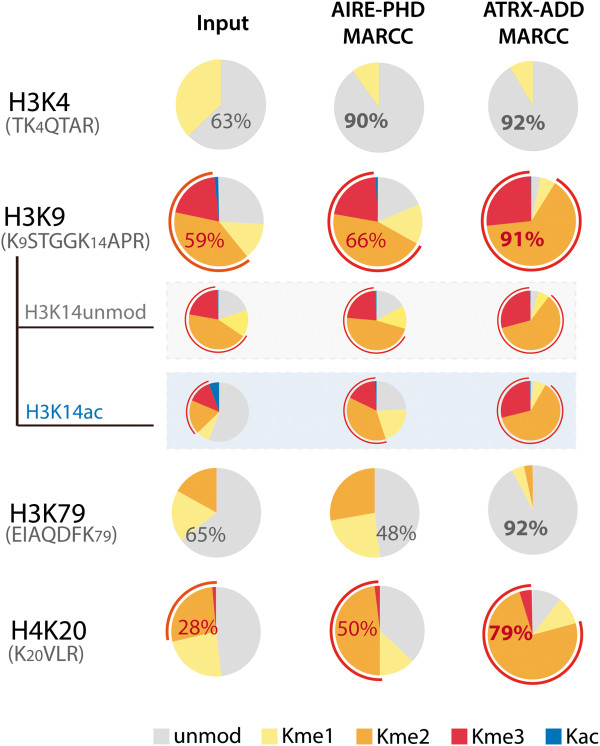
Here we demonstrate an efficient, quantitative, antibody-free, chromatin immunoprecipitation-less (ChIP-less) method for interrogating diverse epigenetic states. At the heart of the workflow are recombinant chromatin reader domains, which target distinct chromatin states with combinatorial PTM patterns. Utilizing a newly designed combinatorial histone peptide microarray, we showed that three reader domains (ATRX-ADD, ING2-PHD and AIRE-PHD) displayed greater specificity towards combinatorial PTM patterns than corresponding commercial histone antibodies. Such specific recognitions were employed to develop a chromatin reader-based affinity enrichment platform (matrix-assisted reader chromatin capture, or MARCC). We successfully applied the reader-based platform to capture unique chromatin states, which were quantitatively profiled by mass spectrometry to reveal interconnections between nucleosomal histone PTMs. Specifically, a highly enriched signature that harbored H3K4me0, H3K9me2/3, H3K79me0 and H4K20me2/3 within the same nucleosome was identified from chromatin enriched by ATRX-ADD. This newly reported PTM combination was enriched in heterochromatin, as revealed by the associated DNA.
Our results suggest the broad utility of recombinant reader domains as an enrichment tool specific to combinatorial PTM patterns, which are difficult to probe directly by antibody-based approaches. The reader affinity platform is compatible with several downstream analyses to investigate the physical coexistence of nucleosomal PTM states associated with specific genomic loci. Collectively, the reader-based workflow will greatly facilitate our understanding of how distinct chromatin states and reader domains function in gene regulatory mechanisms.
组蛋白翻译后修饰(PTMs)是染色质相关过程中的关键表观遗传调控因子。越来越多的证据表明,染色质组蛋白中存在大量的 PTM 组合。这些复杂的模式,而不是单个 PTM,被认为定义了功能性染色质状态。然而,由于缺乏直接的分子工具,在核小体水平上检测组合组蛋白 PTM 模式的能力受到了限制。
在这里,我们展示了一种高效、定量、无抗体、无染色质免疫沉淀(ChIP)的方法,用于检测多种表观遗传状态。该工作流程的核心是重组染色质读取器结构域,这些结构域针对具有组合 PTM 模式的不同染色质状态。我们利用新设计的组合组蛋白肽微阵列,表明三个读取器结构域(ATRX-ADD、ING2-PHD 和 AIRE-PHD)对组合 PTM 模式的特异性高于相应的商业组蛋白抗体。这种特异性识别被用于开发基于染色质读取器的亲和富集平台(基质辅助读取器染色质捕获或 MARCC)。我们成功地将基于读取器的平台应用于捕获独特的染色质状态,这些状态通过质谱进行定量分析,以揭示核小体组蛋白 PTM 之间的相互关系。具体来说,从 ATRX-ADD 富集的染色质中鉴定出一个高度富集的特征,该特征在同一个核小体中含有 H3K4me0、H3K9me2/3、H3K79me0 和 H4K20me2/3。这种新报道的 PTM 组合在异染色质中富集,这是由相关 DNA 揭示的。
我们的结果表明,重组读取器结构域作为一种针对组合 PTM 模式的富集工具具有广泛的应用潜力,而这种组合 PTM 模式很难通过抗体方法直接探测。读取器亲和平台与几种下游分析兼容,可用于研究与特定基因组位点相关的核小体 PTM 状态的物理共存。总的来说,基于读取器的工作流程将极大地促进我们对不同染色质状态和读取器结构域如何在基因调控机制中发挥作用的理解。