Bouibes A, Zaoui A
LGCgE, Polytech'Lille, University of Lille1. Cite Scientifique, Avenue Paul Langevin, 59655 Villeneuve d'Ascq, France.
Sci Rep. 2014 Jun 4;4:5172. doi: 10.1038/srep05172.
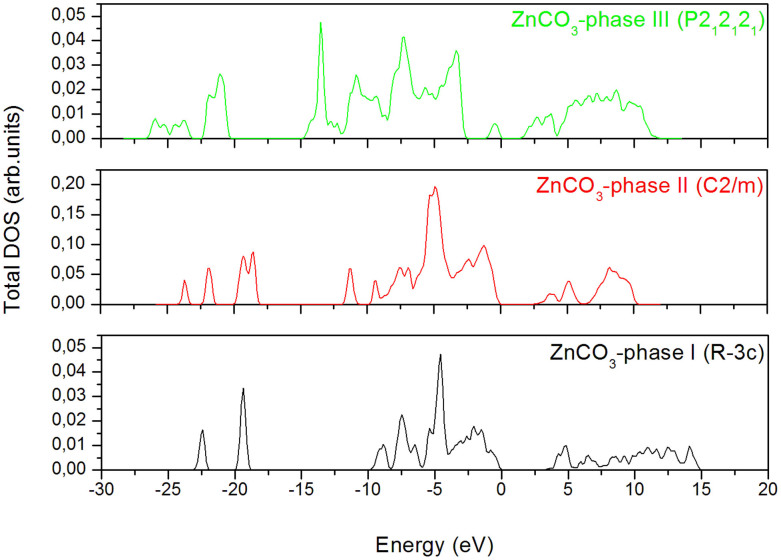
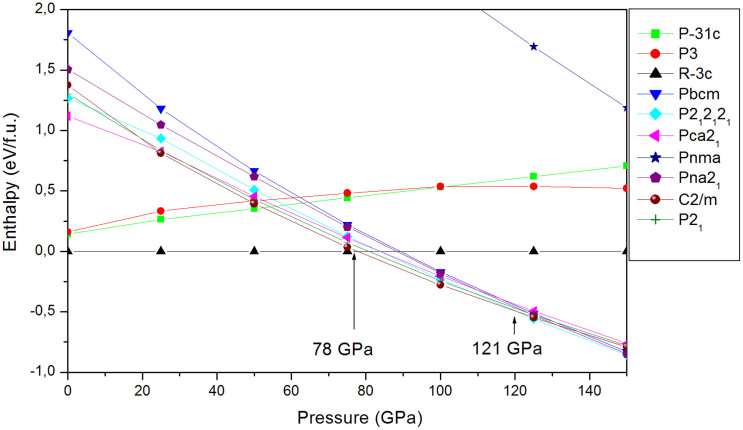
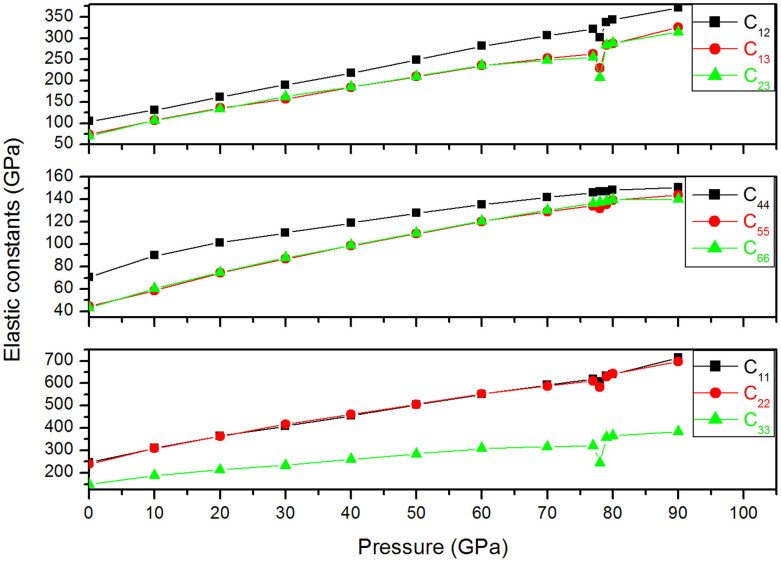
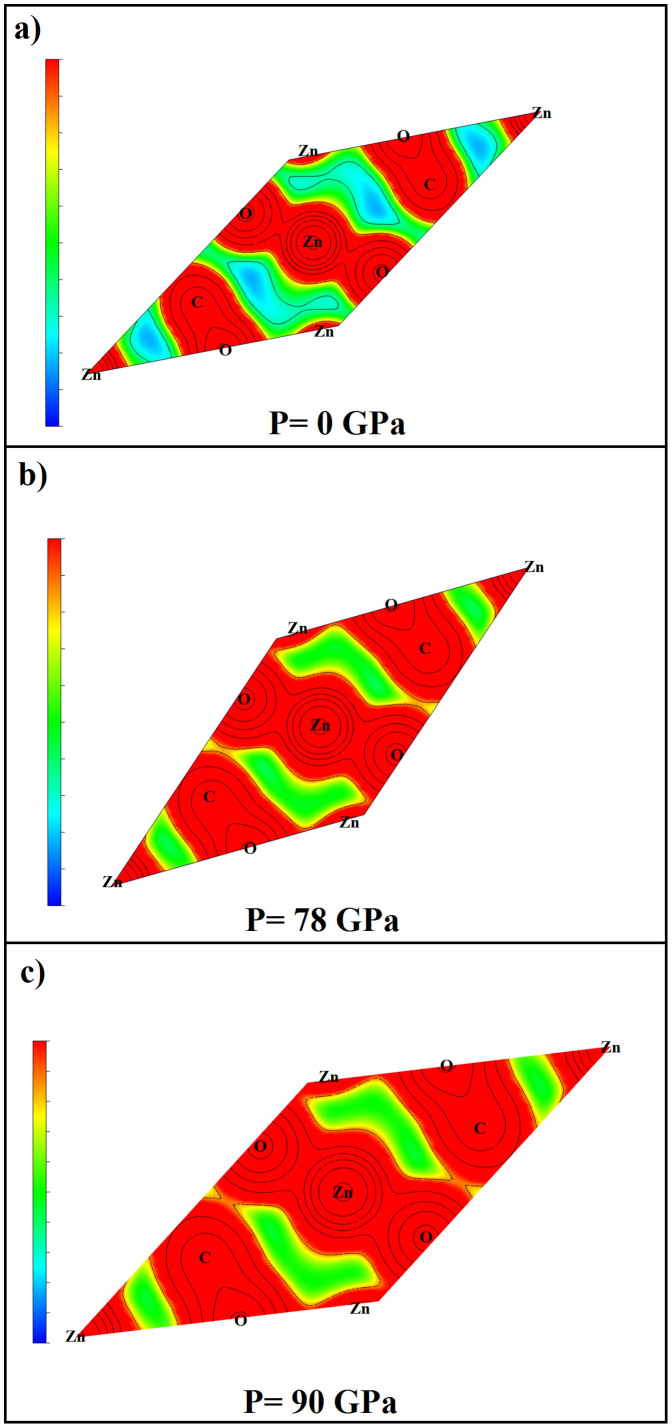
The high-pressure behavior of zinc carbonate ZnCO3 has been investigated using universal structure prediction method together with the density functional theory. In order to explore all possible structures under pressure, separate calculations at high pressure are done here with increasing number of formula units in the unit cell. Two pressures induced phase transitions were considered. The first one occurs at 78 GPa and the second one at 121 GPa. The most stable ZnCO3 at ambient condition corresponds to the space group R-3c (phase I), which is in favorable agreement with experiment. The structure with C2/m space group (phase II) becomes stable between 78 GPa and 121 GPa. Finally, the structure with the space group P2(1)2(1)2(1) (phase III) becomes the most stable when the pressure achieves 121 GPa. Some mechanical properties of R-3c structure were -additionally- calculated and compared with the experimental and previous theoretical data. The resulting behaviors support our findings and confirm the obtained phase transition. Besides, from the analysis of the electronic charge density it comes that at 78 GPa, new bond between oxygen and zinc is formed, what is likely the main cause behind the phase transition.
利用通用结构预测方法结合密度泛函理论研究了碳酸锌(ZnCO₃)的高压行为。为了探索高压下所有可能的结构,本文通过增加晶胞中化学式单元的数量来进行高压下的单独计算。考虑了两个压力诱导的相变。第一个相变发生在78 GPa,第二个相变发生在121 GPa。在环境条件下最稳定的ZnCO₃对应于空间群R-3c(相I),这与实验结果非常吻合。具有C2/m空间群的结构(相II)在78 GPa至121 GPa之间变得稳定。最后,当压力达到121 GPa时,具有空间群P2(1)2(1)2(1)的结构(相III)变得最稳定。此外,计算了R-3c结构的一些力学性能,并与实验数据和先前的理论数据进行了比较。所得行为支持了我们的发现,并证实了所获得的相变。此外,通过对电子电荷密度的分析发现,在78 GPa时,氧和锌之间形成了新的键,这可能是相变背后的主要原因。