Howard Hughes Medical Institute, Program in Cellular and Molecular Medicine, Boston Children's Hospital; and Department of Genetics, Harvard Medical School, Boston, Massachusetts.
Howard Hughes Medical Institute, Program in Cellular and Molecular Medicine, Boston Children's Hospital; and Department of Genetics, Harvard Medical School, Boston, Massachusetts
Cancer Immunol Res. 2014 Sep;2(9):857-66. doi: 10.1158/2326-6066.CIR-14-0090. Epub 2014 Jun 9.
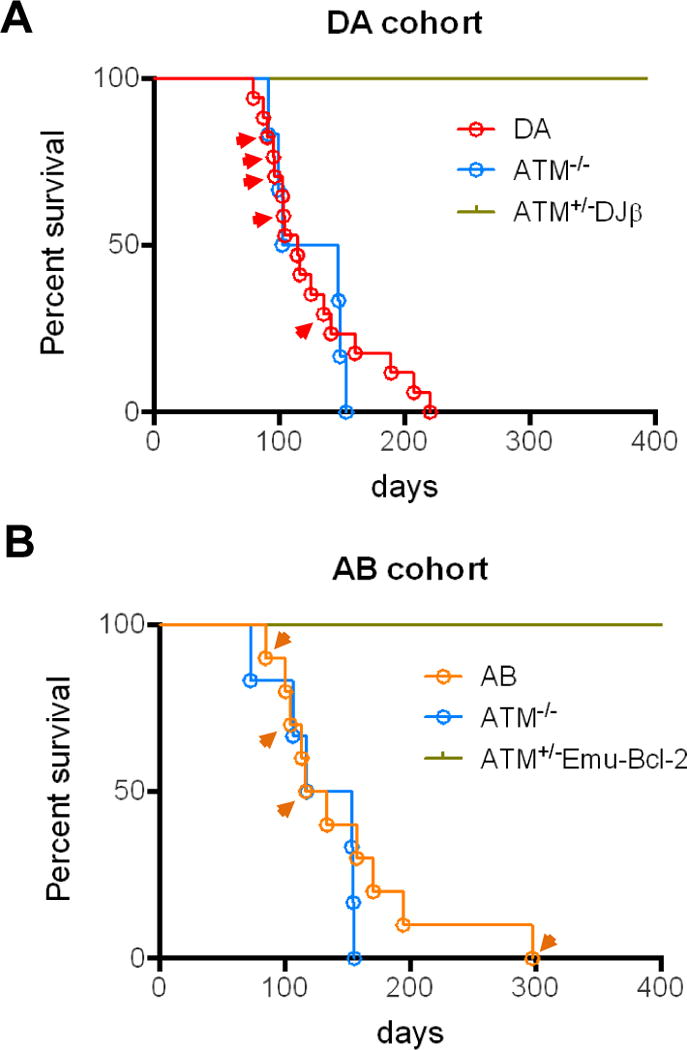
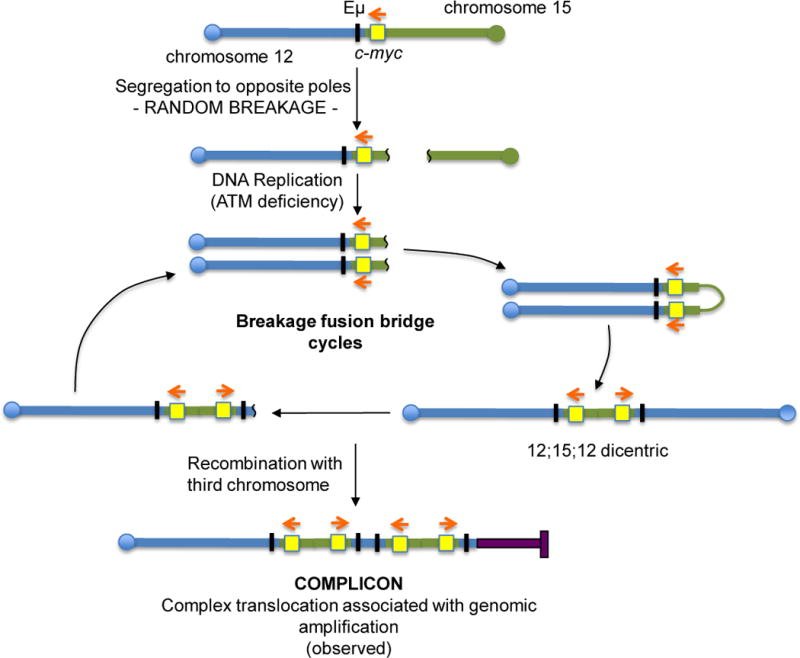
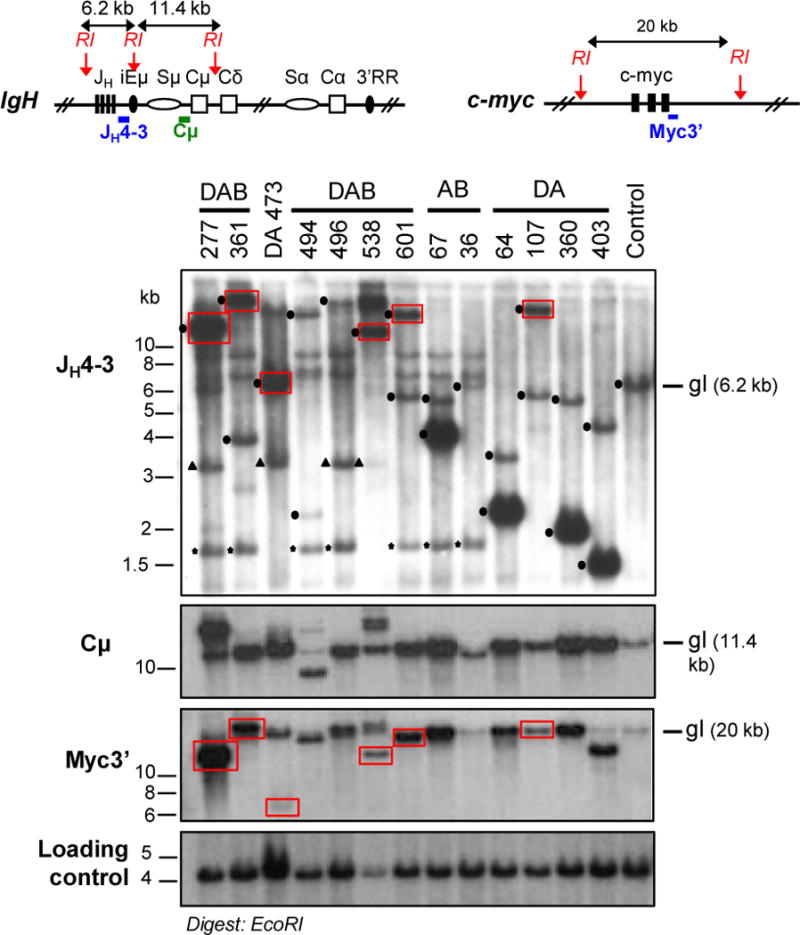
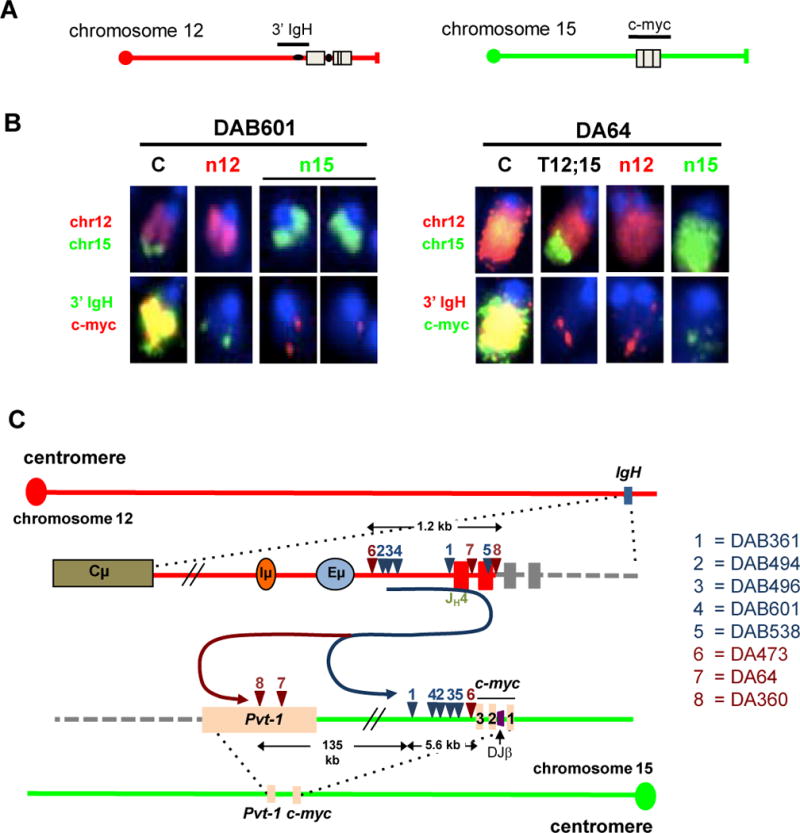
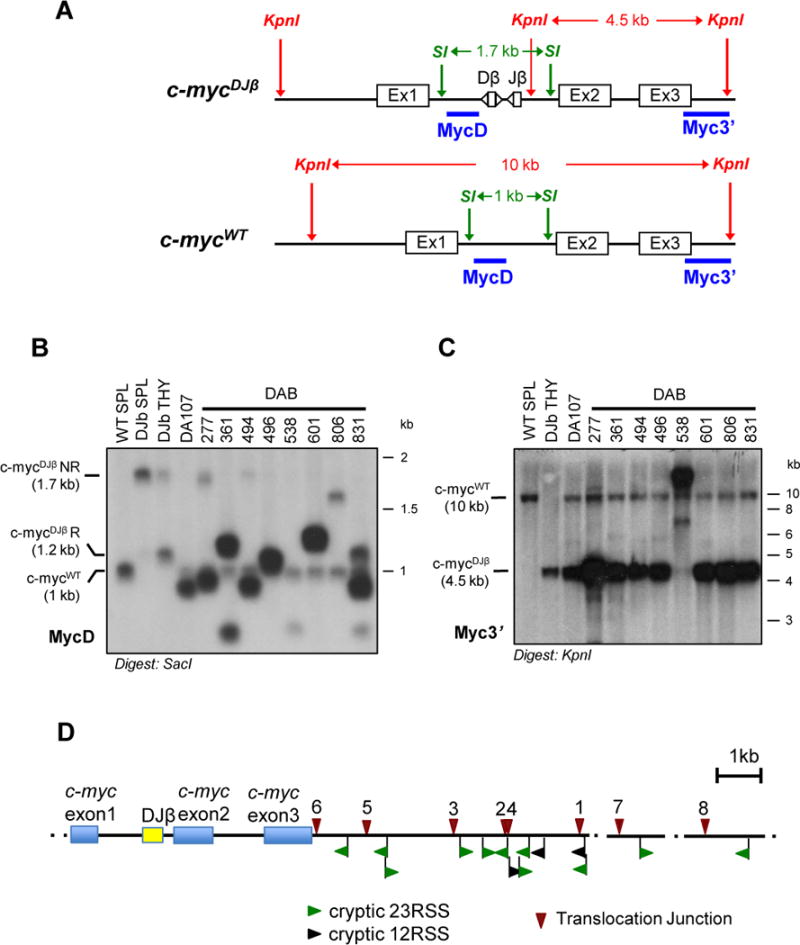
The Ataxia Telangiectasia-mutated (ATM) kinase senses DNA double-strand breaks (DSB) and facilitates their repair. In humans, ATM deficiency predisposes to B- and T-cell lymphomas, but in mice it leads only to thymic lymphomas. We tested the hypothesis that increased DSB frequency at a cellular oncogene could promote B-cell lymphoma by generating ATM-deficient mice with a V(D)J recombination target (DJβ cassette) within c-myc intron 1 ("DA" mice). We also generated ATM-deficient mice carrying an Eμ-Bcl-2 transgene (AB mice) to test whether enhanced cellular survival could promote B-cell lymphomas. About 30% of DA or AB mice and nearly 100% of mice harboring the combined genotypes (DAB mice) developed mature B-cell lymphomas. In all genotypes, B-cell tumors harbored oncogenic c-myc amplification generated by breakage-fusion-bridge (BFB) from dicentric chromosomes formed through fusion of IgH V(D)J recombination-associated DSBs on chromosome 12 to sequences downstream of c-myc on chromosome 15. AB tumors demonstrate that B lineage cells harboring spontaneous DSBs leading to IgH/c-myc dicentrics are blocked from progressing to B-cell lymphomas by cellular apoptotic responses. DA and DAB tumor translocations were strictly linked to the cassette, but occurred downstream, frequently in a 6-kb region adjacent to c-myc that harbors multiple cryptic V(D)J recombination targets, suggesting that bona fide V(D)J target sequences may activate linked cryptic targets. Our findings indicate that ATM deficiency allows IgH V(D)J recombination DSBs in developing B cells to generate dicentric translocations that, via BFB cycles, lead to c-myc-activating oncogenic translocations and amplifications in mature B cells.
共济失调毛细血管扩张症突变(ATM)激酶可感知 DNA 双链断裂(DSB)并促进其修复。在人类中,ATM 缺陷易导致 B 和 T 细胞淋巴瘤,但在小鼠中仅导致胸腺淋巴瘤。我们通过在 c-myc 内含子 1 中产生 ATM 缺陷小鼠的 V(D)J 重组靶标(DJβ 盒)(“DA”小鼠),测试了在细胞癌基因中增加 DSB 频率可通过促进 B 细胞淋巴瘤的假设。我们还生成了携带 Eμ-Bcl-2 转基因的 ATM 缺陷小鼠(AB 小鼠),以测试增强细胞存活是否可以促进 B 细胞淋巴瘤。大约 30%的 DA 或 AB 小鼠和约 100%携带联合基因型(DAB 小鼠)的小鼠发展为成熟的 B 细胞淋巴瘤。在所有基因型中,B 细胞肿瘤均携带通过易位染色体上的 IgH V(D)J 重组相关的 DSB 断裂-融合-桥(BFB)形成的双中心染色体产生的致癌性 c-myc 扩增,这些 DSB 位于 12 号染色体上,靠近 c-myc 的下游。AB 肿瘤表明,通过细胞凋亡反应阻止了带有自发 DSB 的 B 谱系细胞进展为 B 细胞淋巴瘤,这些 DSB 导致 IgH/c-myc 双中心。DA 和 DAB 肿瘤易位严格与盒相关,但发生在下游,经常在靠近 c-myc 的 6kb 区域中,该区域含有多个隐藏的 V(D)J 重组靶标,这表明真正的 V(D)J 靶标序列可能激活连接的隐藏靶标。我们的研究结果表明,ATM 缺陷使发育中的 B 细胞中的 IgH V(D)J 重组 DSB 产生双中心易位,通过 BFB 循环,导致 c-myc 激活的致癌易位和成熟 B 细胞中的扩增。