English Adam C, Salerno William J, Reid Jeffrey G
Human Genome Sequencing Center at Baylor College of Medicine, One Baylor Plaza, Houston 77030, Texas, USA.
BMC Bioinformatics. 2014 Jun 10;15:180. doi: 10.1186/1471-2105-15-180.
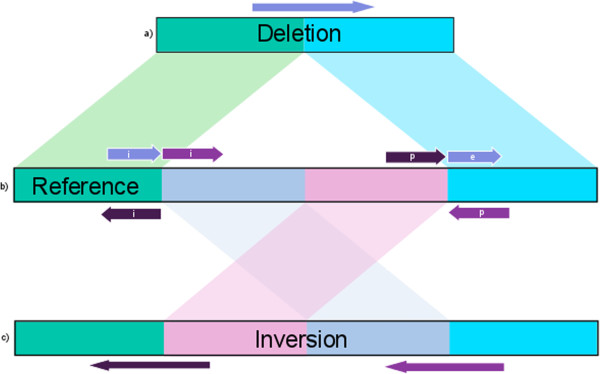
As resequencing projects become more prevalent across a larger number of species, accurate variant identification will further elucidate the nature of genetic diversity and become increasingly relevant in genomic studies. However, the identification of larger genomic variants via DNA sequencing is limited by both the incomplete information provided by sequencing reads and the nature of the genome itself. Long-read sequencing technologies provide high-resolution access to structural variants often inaccessible to shorter reads.
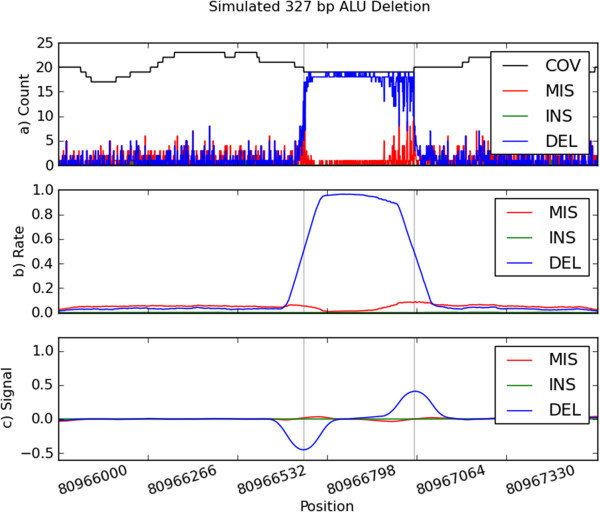
We present PBHoney, software that considers both intra-read discordance and soft-clipped tails of long reads (>10,000 bp) to identify structural variants. As a proof of concept, we identify four structural variants and two genomic features in a strain of Escherichia coli with PBHoney and validate them via de novo assembly. PBHoney is available for download at http://sourceforge.net/projects/pb-jelly/.
Implementing two variant-identification approaches that exploit the high mappability of long reads, PBHoney is demonstrated as being effective at detecting larger structural variants using whole-genome Pacific Biosciences RS II Continuous Long Reads. Furthermore, PBHoney is able to discover two genomic features: the existence of Rac-Phage in isolate; evidence of E. coli's circular genome.
随着重测序项目在越来越多的物种中变得更加普遍,准确的变异识别将进一步阐明遗传多样性的本质,并在基因组研究中变得越来越重要。然而,通过DNA测序识别较大的基因组变异受到测序读数提供的不完整信息以及基因组本身性质的限制。长读长测序技术能够高分辨率地获取短读长通常无法触及的结构变异。
我们展示了PBHoney软件,该软件通过考虑长读长(>10,000 bp)的读内不一致性和软剪切末端来识别结构变异。作为概念验证,我们使用PBHoney在一株大肠杆菌中识别出四个结构变异和两个基因组特征,并通过从头组装对它们进行了验证。PBHoney可从http://sourceforge.net/projects/pb-jelly/下载。
PBHoney实施了两种利用长读长高可映射性的变异识别方法,经证明它能有效地使用全基因组太平洋生物科学公司RS II连续长读长检测更大的结构变异。此外,PBHoney能够发现两个基因组特征:分离株中Rac噬菌体的存在;大肠杆菌环形基因组的证据。