Ratan Aakrosh, Olson Thomas L, Loughran Thomas P, Miller Webb
Center for Comparative Genomics and Bioinformatics, Pennsylvania State University, 506, Wartik Laboratory, University Park, PA, 16802, USA.
Department of Public Health Sciences and Center for Public Health Genomics, University of Virginia, Charlottesville, VA, 22908, USA.
BMC Bioinformatics. 2015 Feb 13;16(1):42. doi: 10.1186/s12859-015-0483-6.
The discovery and mapping of genomic variants is an essential step in most analysis done using sequencing reads. There are a number of mature software packages and associated pipelines that can identify single nucleotide polymorphisms (SNPs) with a high degree of concordance. However, the same cannot be said for tools that are used to identify the other types of variants. Indels represent the second most frequent class of variants in the human genome, after single nucleotide polymorphisms. The reliable detection of indels is still a challenging problem, especially for variants that are longer than a few bases.
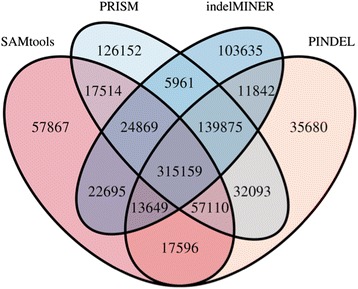
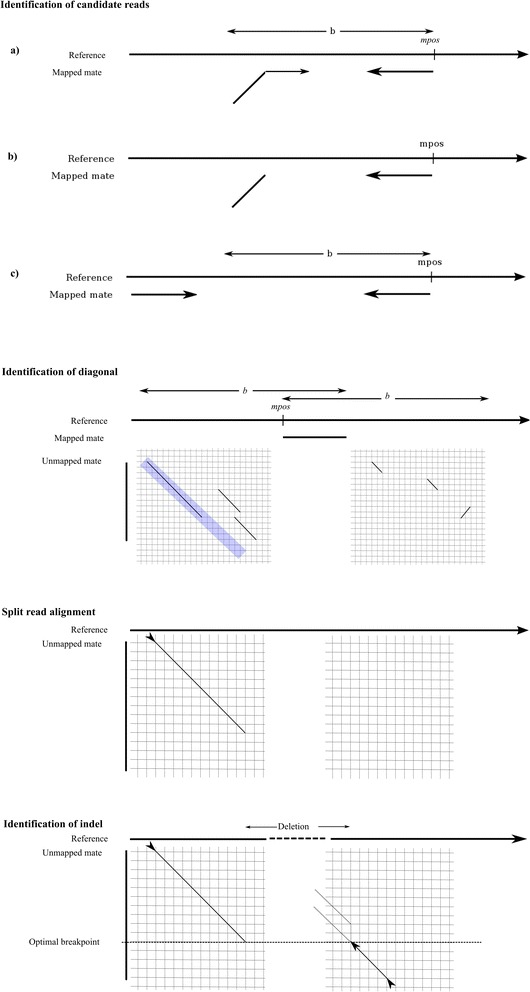
We have developed a set of algorithms and heuristics collectively called indelMINER to identify indels from whole genome resequencing datasets using paired-end reads. indelMINER uses a split-read approach to identify the precise breakpoints for indels of size less than a user specified threshold, and supplements that with a paired-end approach to identify larger variants that are frequently missed with the split-read approach. We use simulated and real datasets to show that an implementation of the algorithm performs favorably when compared to several existing tools.
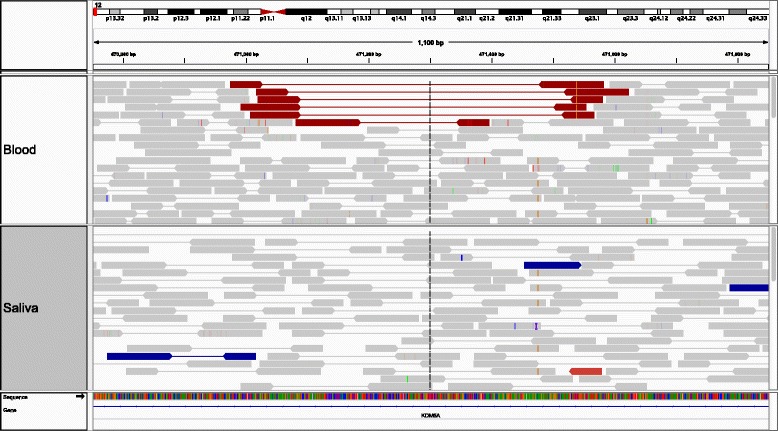
indelMINER can be used effectively to identify indels in whole-genome resequencing projects. The output is provided in the VCF format along with additional information about the variant, including information about its presence or absence in another sample. The source code and documentation for indelMINER can be freely downloaded from www.bx.psu.edu/miller_lab/indelMINER.tar.gz .
基因组变异的发现与定位是大多数基于测序 reads 进行的分析中的关键步骤。有许多成熟的软件包及相关流程能够高度一致地识别单核苷酸多态性(SNP)。然而,用于识别其他类型变异的工具却并非如此。插入缺失(Indel)是人类基因组中仅次于单核苷酸多态性的第二大常见变异类型。可靠地检测插入缺失仍然是一个具有挑战性的问题,尤其是对于长度超过几个碱基的变异。
我们开发了一组统称为 indelMINER 的算法和启发式方法,用于使用双末端 reads 从全基因组重测序数据集中识别插入缺失。indelMINER 使用分裂 reads 方法来识别大小小于用户指定阈值的插入缺失的精确断点,并辅以双末端方法来识别分裂 reads 方法经常遗漏的较大变异。我们使用模拟和真实数据集表明,与几个现有工具相比,该算法的实现表现良好。
indelMINER 可有效地用于全基因组重测序项目中识别插入缺失。输出以 VCF 格式提供,并附带有关变异的其他信息,包括其在另一个样本中是否存在的信息。indelMINER 的源代码和文档可从 www.bx.psu.edu/miller_lab/indelMINER.tar.gz 免费下载。