Department of Paediatrics and Adolescent Medicine, LKS Faculty of Medicine, The University of Hong Kong, 5 Sassoon Road, Hong Kong, China.
BMC Genomics. 2014 Jun 10;15(1):455. doi: 10.1186/1471-2164-15-455.
Predicting the functional impact of amino acid substitutions (AAS) caused by nonsynonymous single nucleotide polymorphisms (nsSNPs) is becoming increasingly important as more and more novel variants are being discovered. Bioinformatics analysis is essential to predict potentially causal or contributing AAS to human diseases for further analysis, as for each genome, thousands of rare or private AAS exist and only a very small number of which are related to an underlying disease. Existing algorithms in this field still have high false prediction rate and novel development is needed to take full advantage of vast amount of genomic data.
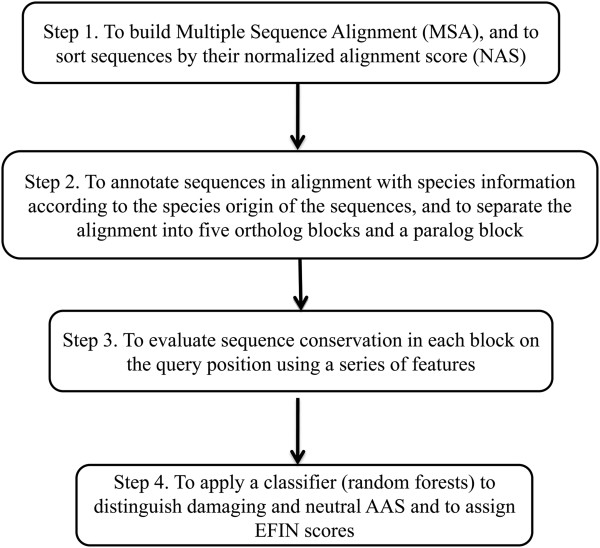
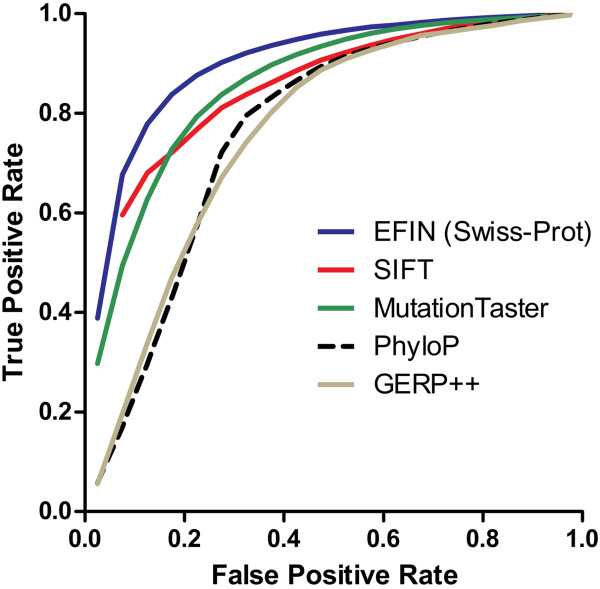
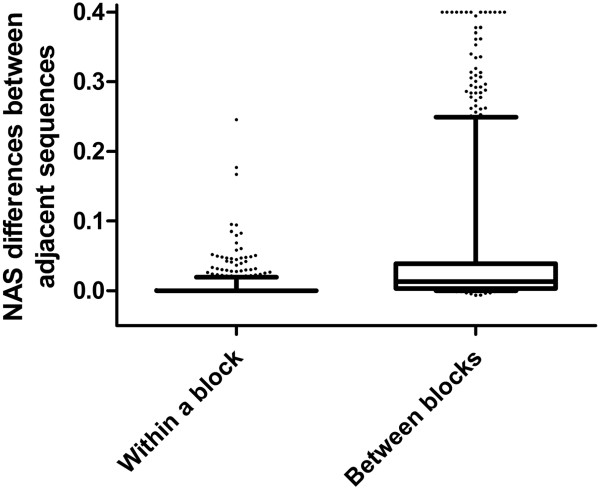
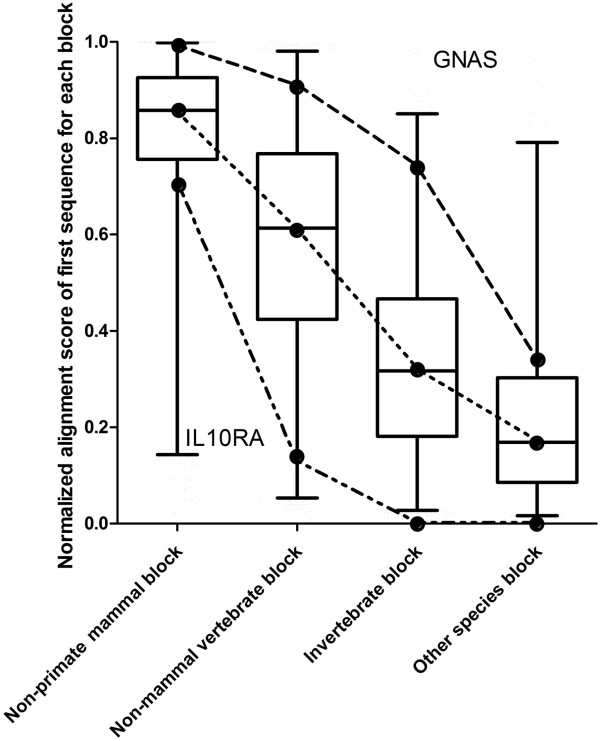
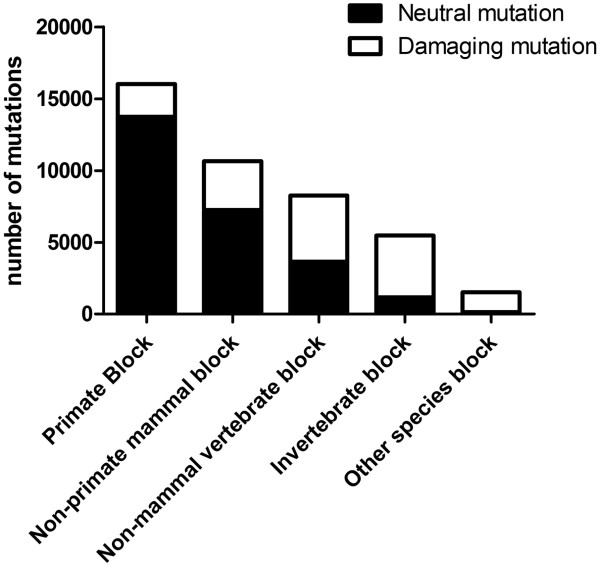
Here we report a novel algorithm that features two innovative changes: 1. making better use of sequence conservation information by grouping the homologous protein sequences into six blocks according to evolutionary distances to human and evaluating sequence conservation in each block independently, and 2. including as many such homologous sequences as possible in analyses. Random forests are used to evaluate sequence conservation in each block and to predict potential impact of an AAS on protein function. Testing of this algorithm on a comprehensive dataset showed significant improvement on prediction accuracy upon currently widely-used programs. The algorithm and a web-based application tool implementing it, EFIN (Evaluation of Functional Impact of Nonsynonymous SNPs) were made freely available (http://paed.hku.hk/efin/) to the public.
Grouping homologous sequences into different blocks according to the evolutionary distance of the species to human and evaluating sequence conservation in each group independently significantly improved prediction accuracy. This approach may help us better understand the roles of genetic variants in human disease and health.
随着越来越多的新型变异被发现,预测由非同义单核苷酸多态性(nsSNP)引起的氨基酸替换(AAS)对功能的影响变得越来越重要。生物信息学分析对于预测可能导致人类疾病的因果或贡献性 AAS 至关重要,因为对于每个基因组,都存在数千种罕见或特定的 AAS,其中只有极少数与潜在疾病有关。该领域现有的算法仍然存在很高的假阳性预测率,因此需要新的开发来充分利用大量的基因组数据。
我们在此报告了一种新的算法,其具有两个创新的变化:1. 通过根据与人类的进化距离将同源蛋白序列分为六个块,更好地利用序列保守性信息,并独立评估每个块中的序列保守性;2. 在分析中尽可能多地包含此类同源序列。随机森林用于评估每个块中的序列保守性,并预测 AAS 对蛋白质功能的潜在影响。在一个综合数据集上对该算法进行测试表明,与当前广泛使用的程序相比,预测准确性有了显著提高。该算法和一个基于网络的应用程序工具 EFIN(非同义 SNP 功能影响评估)已免费向公众提供(http://paed.hku.hk/efin/)。
根据与人类的进化距离将同源序列分为不同的块,并独立评估每个组中的序列保守性,显著提高了预测准确性。这种方法可能有助于我们更好地理解遗传变异在人类疾病和健康中的作用。