Narise Kosuke, Okuda Kensuke, Enomoto Yukihiro, Hirayama Tasuku, Nagasawa Hideko
Laboratory of Pharmaceutical and Medicinal Chemistry, Gifu Pharmaceutical University, Daigaku-nishi, Gifu, Japan.
Drug Des Devel Ther. 2014 Jun 6;8:701-17. doi: 10.2147/DDDT.S59679. eCollection 2014.
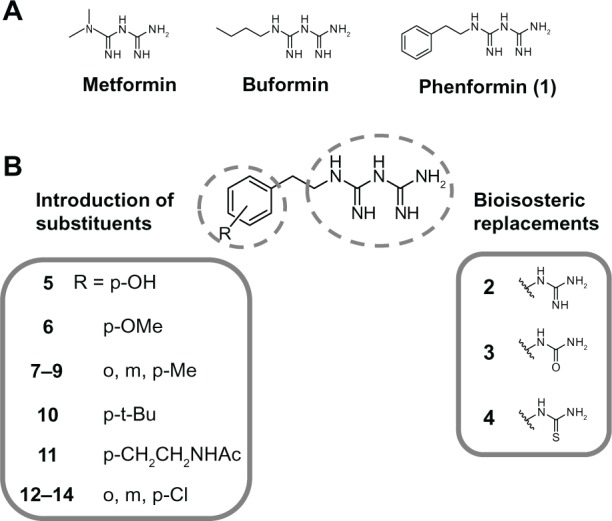
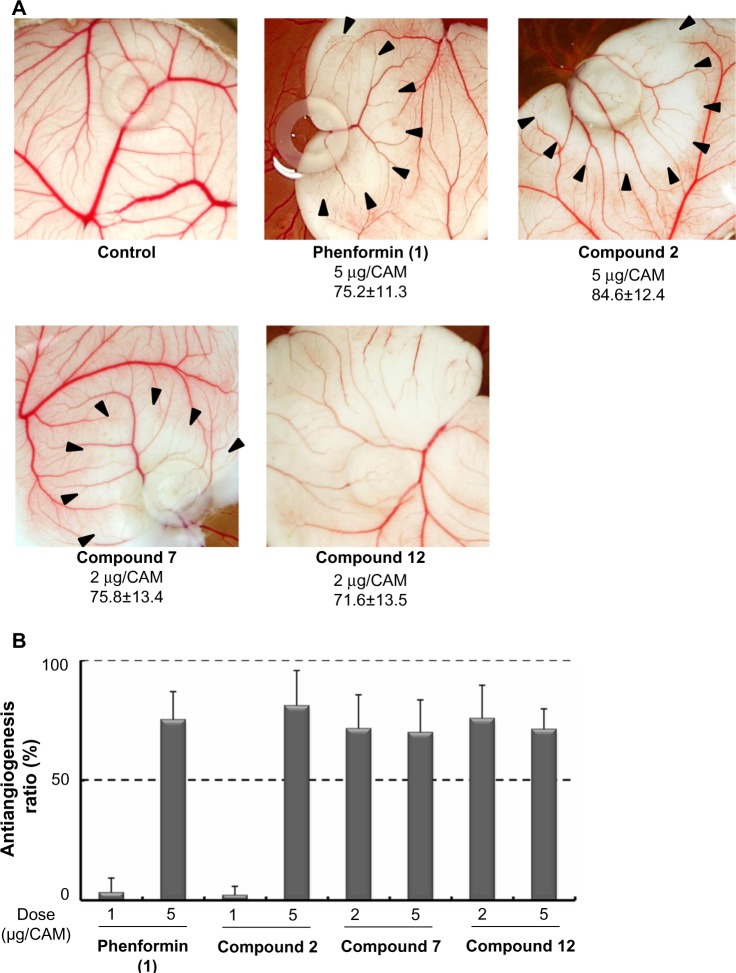
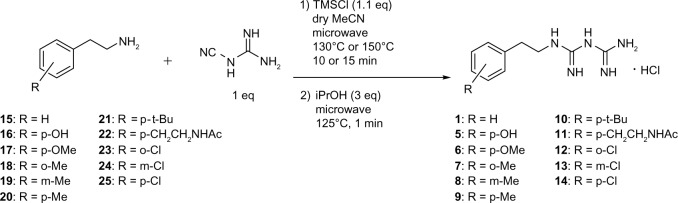
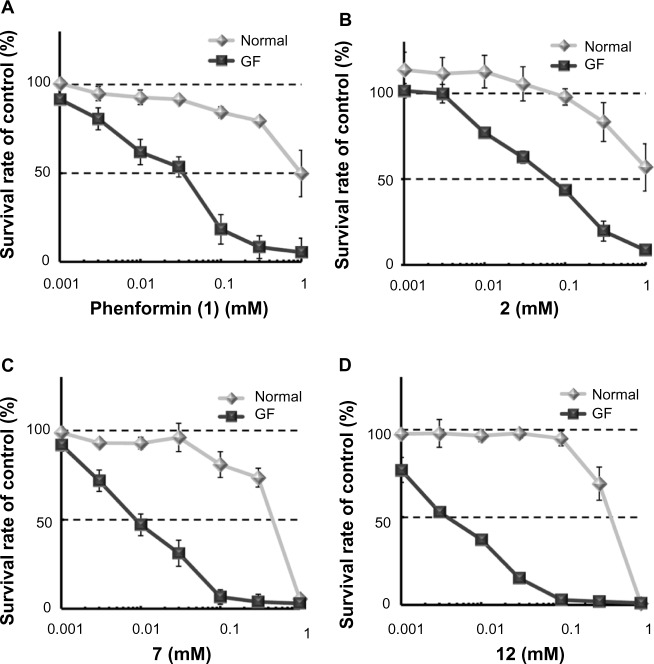
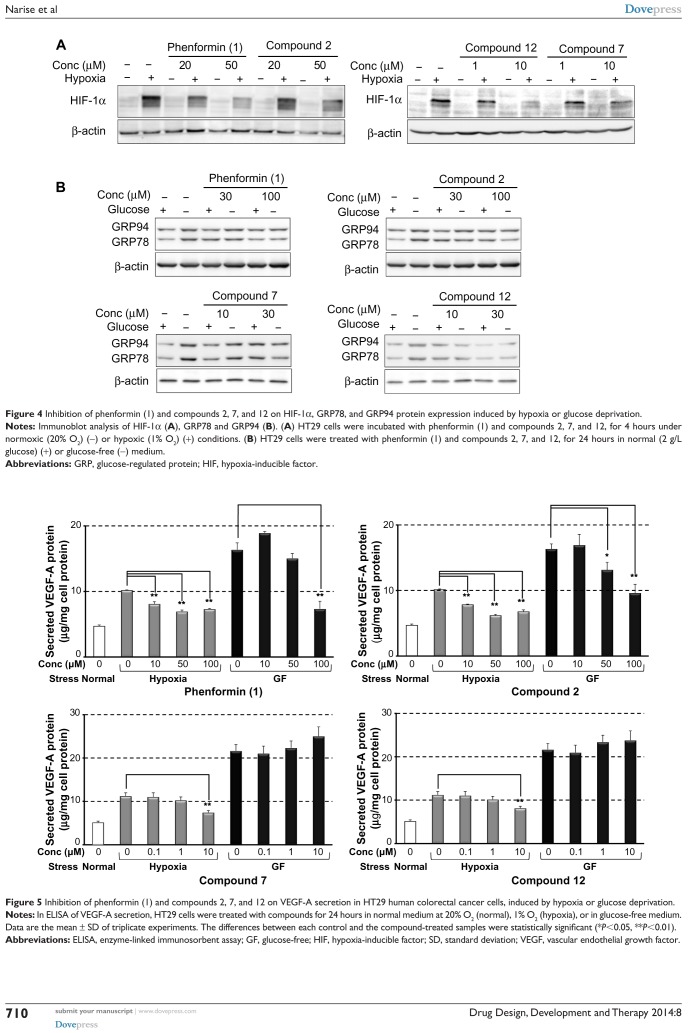
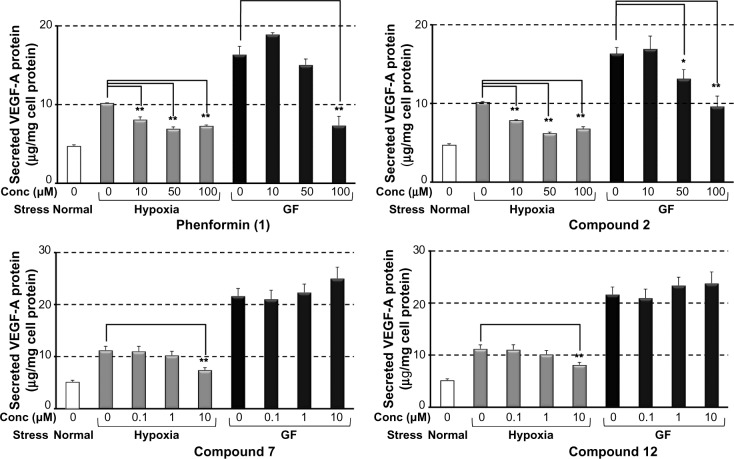
Adaptive cellular responses resulting from multiple microenvironmental stresses, such as hypoxia and nutrient deprivation, are potential novel drug targets for cancer treatment. Accordingly, we focused on developing anticancer agents targeting the tumor microenvironment (TME). In this study, to search for selective antitumor agents blocking adaptive responses in the TME, thirteen new compounds, designed and synthesized on the basis of the arylmethylbiguanide scaffold of phenformin, were used in structure activity relationship studies of inhibition of hypoxia inducible factor (HIF)-1 and unfolded protein response (UPR) activation and of selective cytotoxicity under glucose-deprived stress conditions, using HT29 cells. We conducted luciferase reporter assays using stable cell lines expressing either an HIF-1-responsive reporter gene or a glucose-regulated protein 78 promoter-reporter gene, which were induced by hypoxia and glucose deprivation stress, respectively, to screen for TME-targeting antitumor drugs. The guanidine analog (compound 2), obtained by bioisosteric replacement of the biguanide group, had activities comparable with those of phenformin (compound 1). Introduction of various substituents on the phenyl ring significantly affected the activities. In particular, the o-methylphenyl analog compound 7 and the o-chlorophenyl analog compound 12 showed considerably more potent inhibitory effects on HIF-1 and UPR activation than did phenformin, and excellent selective cytotoxicity under glucose deprivation. These compounds, therefore, represent an improvement over phenformin. They also suppressed HIF-1- and UPR-related protein expression and secretion of vascular endothelial growth factor-A. Moreover, these compounds exhibited significant antiangiogenic effects in the chick chorioallantoic membrane assay. Our structural development studies of biguanide derivatives provided promising candidates for a novel anticancer agent targeting the TME for selective cancer therapy, to be subjected to further in vivo study.
由多种微环境应激(如缺氧和营养剥夺)引发的适应性细胞反应是癌症治疗潜在的新型药物靶点。因此,我们专注于开发针对肿瘤微环境(TME)的抗癌药物。在本研究中,为了寻找能够阻断TME中适应性反应的选择性抗肿瘤药物,基于苯乙双胍的芳基甲基双胍支架设计并合成了13种新化合物,利用HT29细胞对其进行抑制缺氧诱导因子(HIF)-1和未折叠蛋白反应(UPR)激活以及在葡萄糖剥夺应激条件下的选择性细胞毒性的构效关系研究。我们使用分别表达HIF-1反应性报告基因或葡萄糖调节蛋白78启动子-报告基因的稳定细胞系进行荧光素酶报告基因检测,这两种基因分别由缺氧和葡萄糖剥夺应激诱导,以筛选靶向TME的抗肿瘤药物。通过双胍基团的生物电子等排体置换得到的胍类似物(化合物2)具有与苯乙双胍(化合物1)相当的活性。在苯环上引入各种取代基显著影响了活性。特别是,邻甲基苯基类似物化合物7和邻氯苯基类似物化合物12对HIF-1和UPR激活的抑制作用比苯乙双胍更强,并且在葡萄糖剥夺条件下具有出色的选择性细胞毒性。因此,这些化合物是对苯乙双胍的改进。它们还抑制了HIF-1和UPR相关蛋白的表达以及血管内皮生长因子-A的分泌。此外,这些化合物在鸡胚绒毛尿囊膜试验中表现出显著的抗血管生成作用。我们对双胍衍生物的结构开发研究为一种新型靶向TME用于选择性癌症治疗的抗癌药物提供了有前景的候选物,有待进一步进行体内研究。