Vander Griend Donald J, Litvinov Ivan V, Isaacs John T
1. Chemical Therapeutics Program, The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. ; 3. The Brady Urological Institute, Johns Hopkins.
1. Chemical Therapeutics Program, The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. ; 2. Cellular and Molecular Medicine Graduate Program at Johns Hopkins.
Int J Biol Sci. 2014 Jun 10;10(6):627-42. doi: 10.7150/ijbs.8756. eCollection 2014.
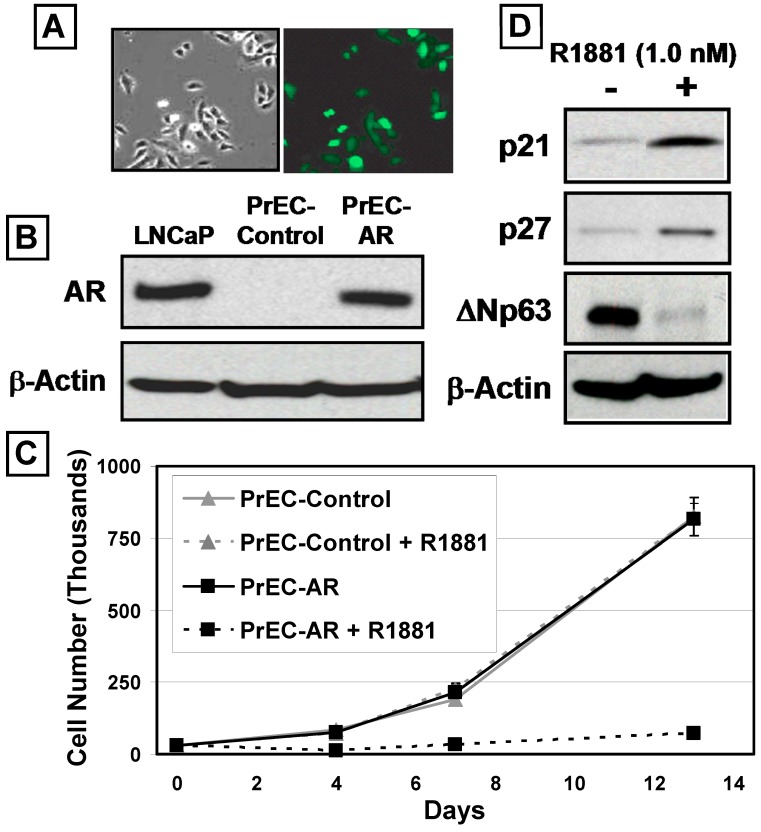
In normal prostate, androgen-dependent androgen receptor (AR) signaling within prostate stromal cells induces their secretion of paracrine factors, termed "andromedins" which stimulate growth of the epithelial cells. The present studies demonstrate that androgen-dependent andromedin-driven growth stimulation is counter-balanced by androgen-induced AR signaling within normal adult prostate epithelial cells resulting in terminal G0 growth arrest coupled with terminal differentiation into ΔNp63-negative, PSA-expressing secretory luminal cells. This cell autonomous AR-driven terminal differentiation requires DNA-binding of the AR protein, is associated with decreases in c-Myc m-RNA and protein, are coupled with increases in p21, p27, and SKP-2 protein expression, and does not require functional p53. These changes result in down-regulation of Cyclin D1 protein and RB phosphoryation. shRNA knockdown documents that neither RB, p21, p27 alone or in combination are required for such AR-induced G0 growth arrest. Transgenic expression of a constitutive vector to prevent c-Myc down-regulation overrides AR-mediated growth arrest in normal prostate epithelial cells, which documents that AR-induced c-Myc down-regulation is critical in terminal growth arrest of normal prostate epithelial cells. In contrast, in prostate cancer cells, androgen-induced AR signaling paradoxically up-regulates c-Myc expression and stimulates growth as documented by inhibition of both of these responses following exposure to the AR antagonist, bicalutamide. These data document that AR signaling is converted from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells during prostatic carcinogenesis and that this conversion involves a gain of function for regulation of c-Myc expression.
在正常前列腺中,前列腺基质细胞内雄激素依赖性雄激素受体(AR)信号传导诱导其分泌旁分泌因子,即“雄激素调节素”,刺激上皮细胞生长。目前的研究表明,雄激素依赖性雄激素调节素驱动的生长刺激被正常成年前列腺上皮细胞内雄激素诱导的AR信号传导所抵消,导致终末G0生长停滞,并伴随终末分化为ΔNp63阴性、表达前列腺特异性抗原(PSA)的分泌性腔细胞。这种细胞自主的AR驱动的终末分化需要AR蛋白的DNA结合,与c-Myc mRNA和蛋白的减少相关,与p21、p27和SKP-2蛋白表达的增加相关,并且不需要功能性p53。这些变化导致细胞周期蛋白D1蛋白下调和RB磷酸化。短发夹RNA(shRNA)敲低表明,RB、p21、p27单独或联合使用都不是AR诱导的G0生长停滞所必需的。组成型载体的转基因表达以防止c-Myc下调可克服正常前列腺上皮细胞中AR介导的生长停滞,这表明AR诱导的c-Myc下调在正常前列腺上皮细胞的终末生长停滞中至关重要。相反,在前列腺癌细胞中,雄激素诱导的AR信号传导反常地上调c-Myc表达并刺激生长,这在暴露于AR拮抗剂比卡鲁胺后对这两种反应的抑制中得到证实。这些数据表明,在前列腺癌发生过程中,AR信号传导从正常前列腺上皮细胞中的生长抑制因子转变为前列腺癌细胞中的癌基因,并且这种转变涉及c-Myc表达调节的功能获得。