Marine Genomics Laboratory, Harte Research Institute, Texas A&M University-Corpus Christi , Corpus Christi, TX , USA.
PeerJ. 2014 Jun 10;2:e431. doi: 10.7717/peerj.431. eCollection 2014.
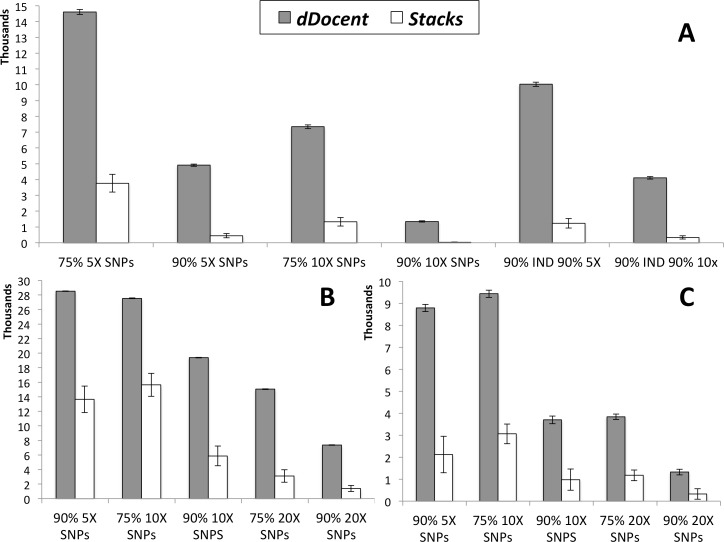
Restriction-site associated DNA sequencing (RADseq) has become a powerful and useful approach for population genomics. Currently, no software exists that utilizes both paired-end reads from RADseq data to efficiently produce population-informative variant calls, especially for non-model organisms with large effective population sizes and high levels of genetic polymorphism. dDocent is an analysis pipeline with a user-friendly, command-line interface designed to process individually barcoded RADseq data (with double cut sites) into informative SNPs/Indels for population-level analyses. The pipeline, written in BASH, uses data reduction techniques and other stand-alone software packages to perform quality trimming and adapter removal, de novo assembly of RAD loci, read mapping, SNP and Indel calling, and baseline data filtering. Double-digest RAD data from population pairings of three different marine fishes were used to compare dDocent with Stacks, the first generally available, widely used pipeline for analysis of RADseq data. dDocent consistently identified more SNPs shared across greater numbers of individuals and with higher levels of coverage. This is due to the fact that dDocent quality trims instead of filtering, incorporates both forward and reverse reads (including reads with INDEL polymorphisms) in assembly, mapping, and SNP calling. The pipeline and a comprehensive user guide can be found at http://dDocent.wordpress.com.
限制性位点相关 DNA 测序(RADseq)已经成为群体基因组学的一种强大而有用的方法。目前,还没有软件可以利用 RADseq 数据的配对末端读取来有效地生成群体信息变异调用,特别是对于具有大有效种群大小和高水平遗传多态性的非模式生物。dDocent 是一个具有用户友好的命令行界面的分析管道,旨在将单独标记的 RADseq 数据(具有双切割位点)处理成有信息的 SNP/Indels,用于群体水平分析。该管道使用 BASH 编写,利用数据缩减技术和其他独立的软件包来执行质量修剪和接头去除、RAD 位点的从头组装、读映射、SNP 和 Indel 调用以及基线数据过滤。使用来自三种不同海洋鱼类的群体配对的双消化 RAD 数据来比较 dDocent 和 Stacks,Stacks 是第一个可用于 RADseq 数据分析的广泛使用的管道。dDocent 始终能够识别更多的 SNP,这些 SNP 在更多的个体中共享,并且具有更高的覆盖水平。这是因为 dDocent 进行质量修剪而不是过滤,在组装、映射和 SNP 调用中同时包含正向和反向读取(包括具有 INDEL 多态性的读取)。该管道和综合用户指南可在 http://dDocent.wordpress.com 上找到。