Division of Bioinformatics and Statistics, School of Public Health, National Defense Medical Center, Taipei 114, Taiwan.
Department of Surgery, Cathay General Hospital, Taipei 106, Taiwan.
Dis Markers. 2014;2014:634123. doi: 10.1155/2014/634123. Epub 2014 May 19.
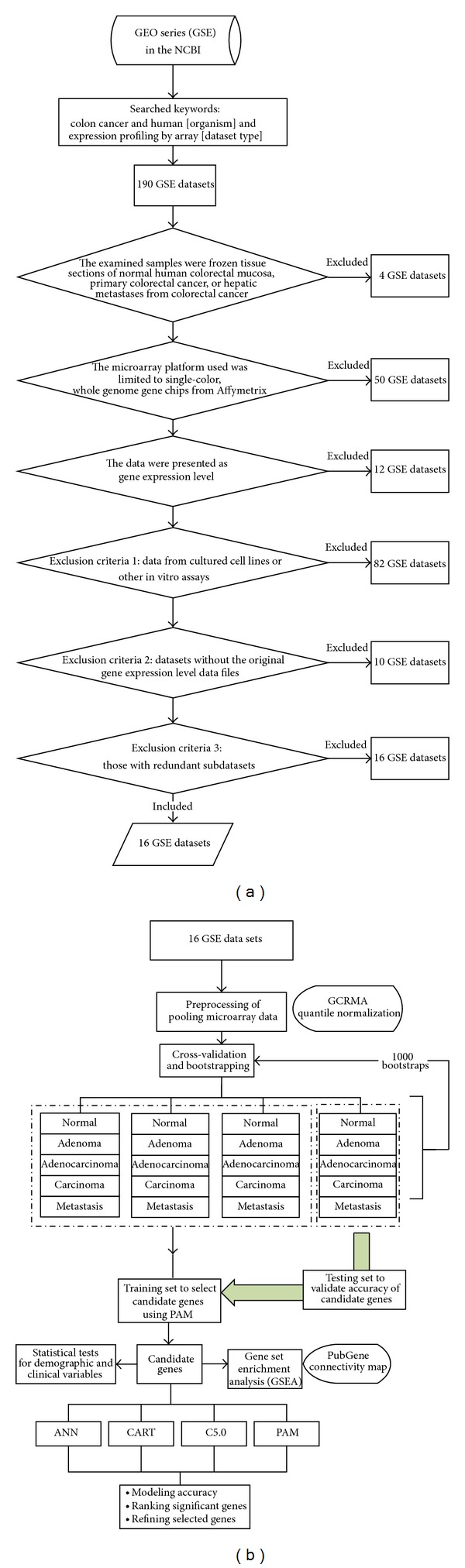
Microarray technology shows great potential but previous studies were limited by small number of samples in the colorectal cancer (CRC) research. The aims of this study are to investigate gene expression profile of CRCs by pooling cDNA microarrays using PAM, ANN, and decision trees (CART and C5.0).
Pooled 16 datasets contained 88 normal mucosal tissues and 1186 CRCs. PAM was performed to identify significant expressed genes in CRCs and models of PAM, ANN, CART, and C5.0 were constructed for screening candidate genes via ranking gene order of significances.
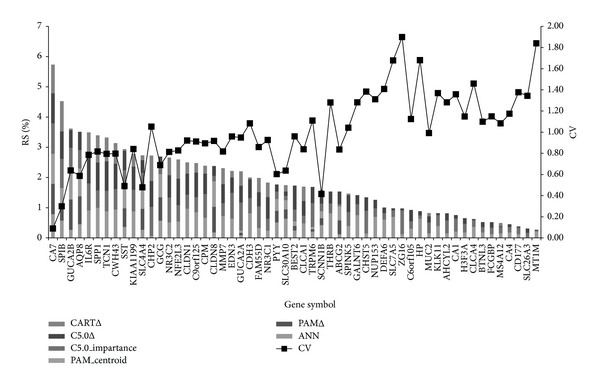
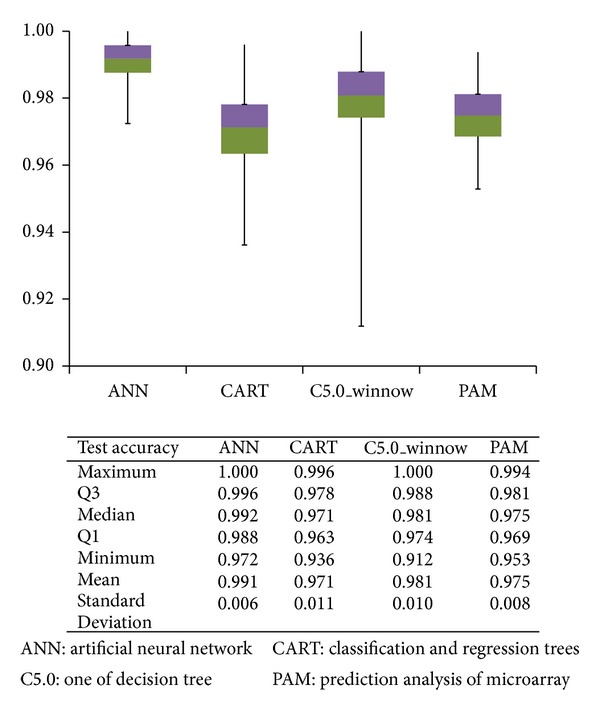
The first screening identified 55 genes. The test accuracy of each model was over 0.97 averagely. Less than eight genes achieve excellent classification accuracy. Combining the results of four models, we found the top eight differential genes in CRCs; suppressor genes, CA7, SPIB, GUCA2B, AQP8, IL6R and CWH43; oncogenes, SPP1 and TCN1. Genes of higher significances showed lower variation in rank ordering by different methods.
We adopted a two-tier genetic screen, which not only reduced the number of candidate genes but also yielded good accuracy (nearly 100%). This method can be applied to future studies. Among the top eight genes, CA7, TCN1, and CWH43 have not been reported to be related to CRC.
微阵列技术显示出巨大的潜力,但以前的研究受到结直肠癌(CRC)研究中样本数量较少的限制。本研究旨在通过使用 PAM、ANN 和决策树(CART 和 C5.0)对 cDNA 微阵列进行汇集,来研究 CRC 的基因表达谱。
汇集了包含 88 个正常黏膜组织和 1186 个 CRC 的 16 个数据集。通过 PAM 鉴定 CRC 中显著表达的基因,并构建 PAM、ANN、CART 和 C5.0 模型,通过对基因显著性排序来筛选候选基因。
第一次筛选确定了 55 个基因。每个模型的测试准确率平均超过 0.97。不到 8 个基因达到了优秀的分类准确率。结合四个模型的结果,我们发现 CRC 中存在前 8 个差异基因:抑癌基因 CA7、SPIB、GUCA2B、AQP8、IL6R 和 CWH43;致癌基因 SPP1 和 TCN1。基因的显著性越高,不同方法的排序变化越小。
我们采用了两阶段遗传筛选,不仅减少了候选基因的数量,而且还获得了接近 100%的良好准确性。这种方法可以应用于未来的研究。在这 8 个基因中,CA7、TCN1 和 CWH43 尚未被报道与 CRC 有关。