Heckman Laura Dean, Chahrour Maria H, Zoghbi Huda Y
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, United States.
Division of Genetics, Department of Medicine, Harvard Medical School, Boston, United States.
Elife. 2014 Jun 26;3:e02676. doi: 10.7554/eLife.02676.
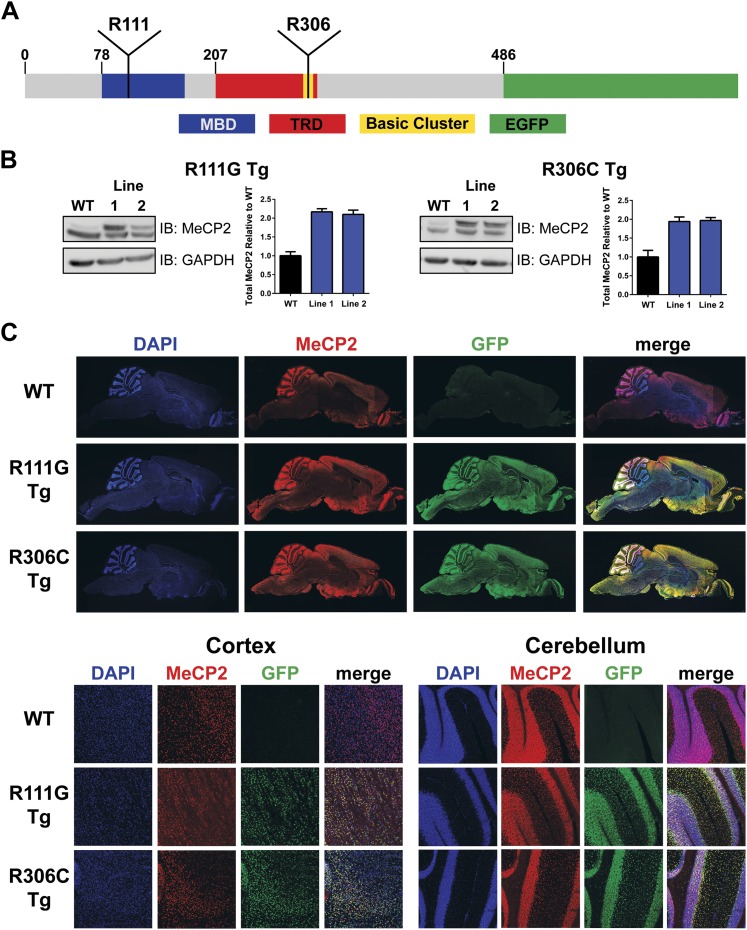
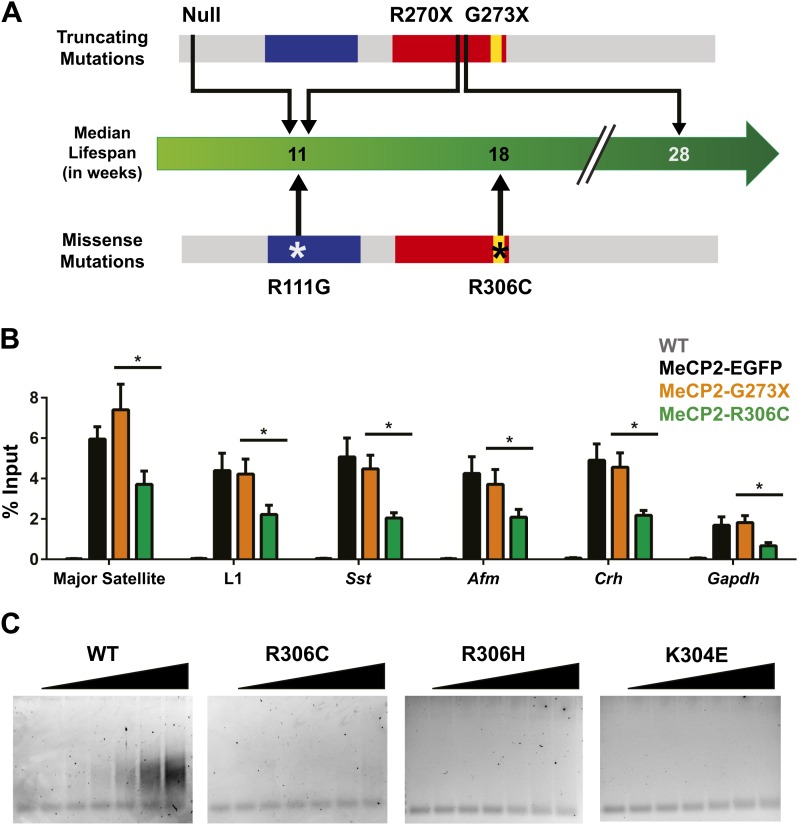
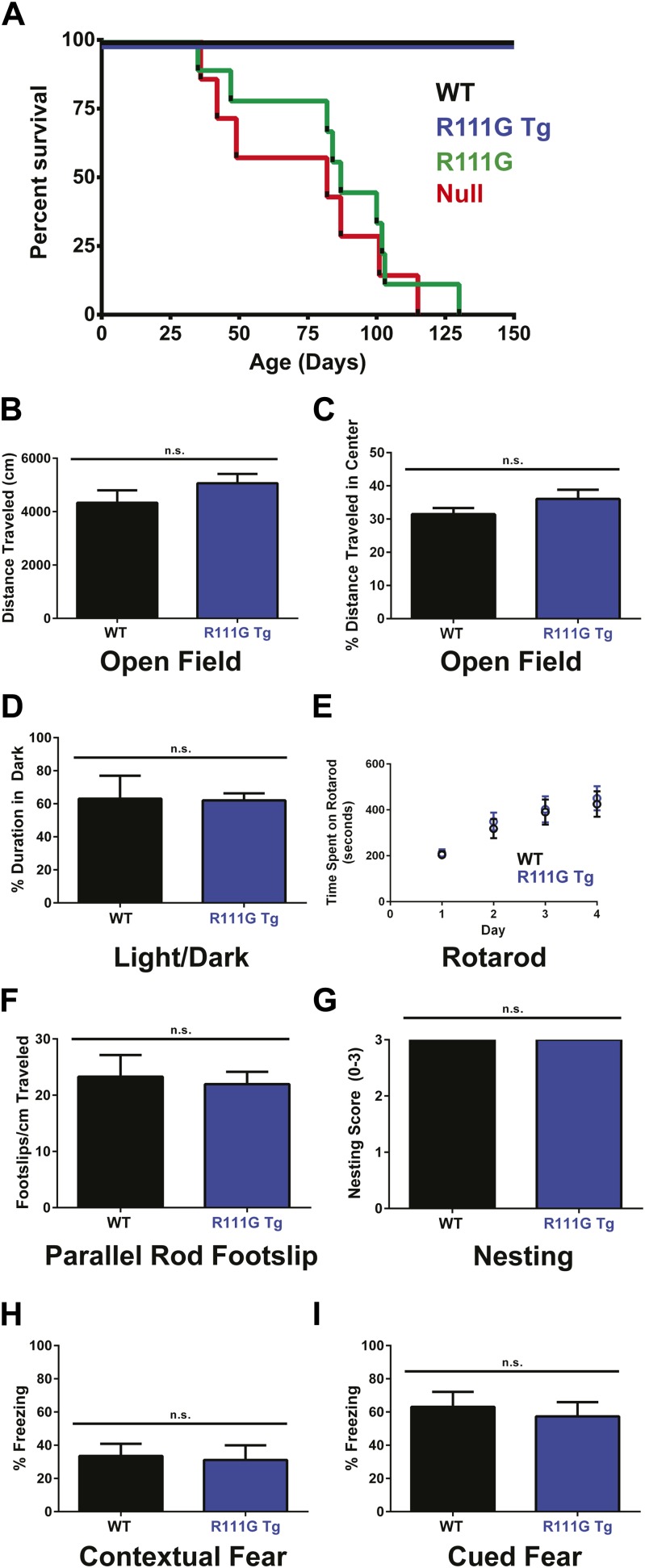
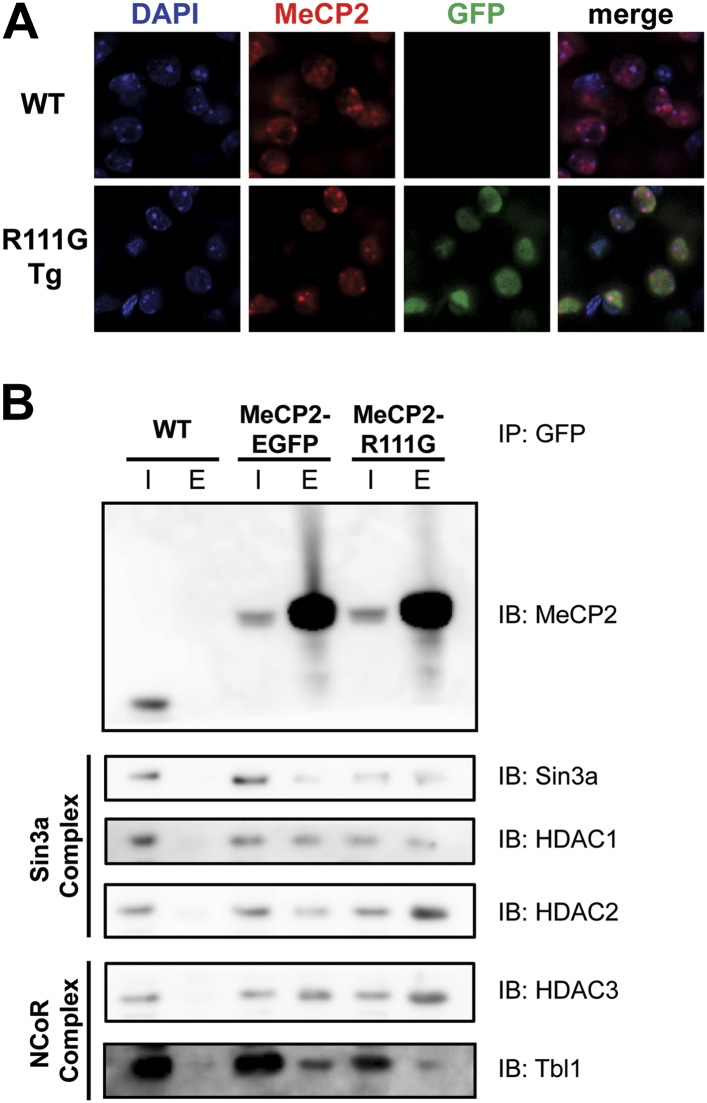
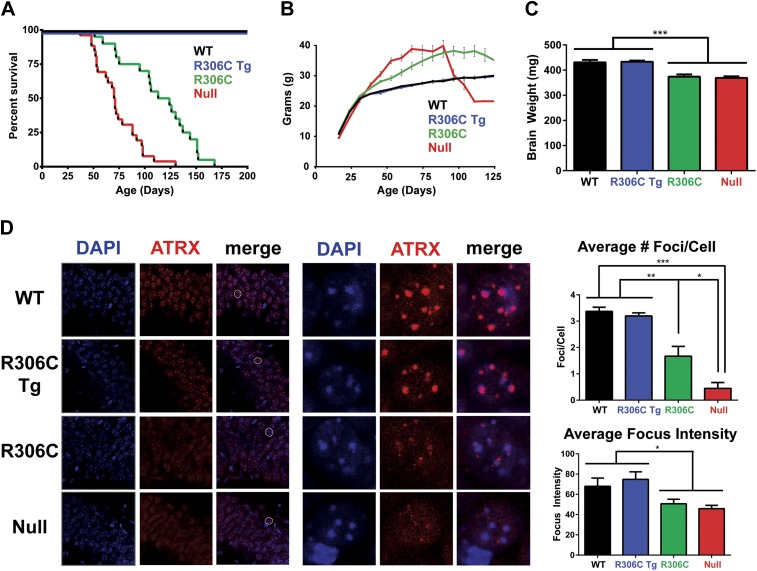
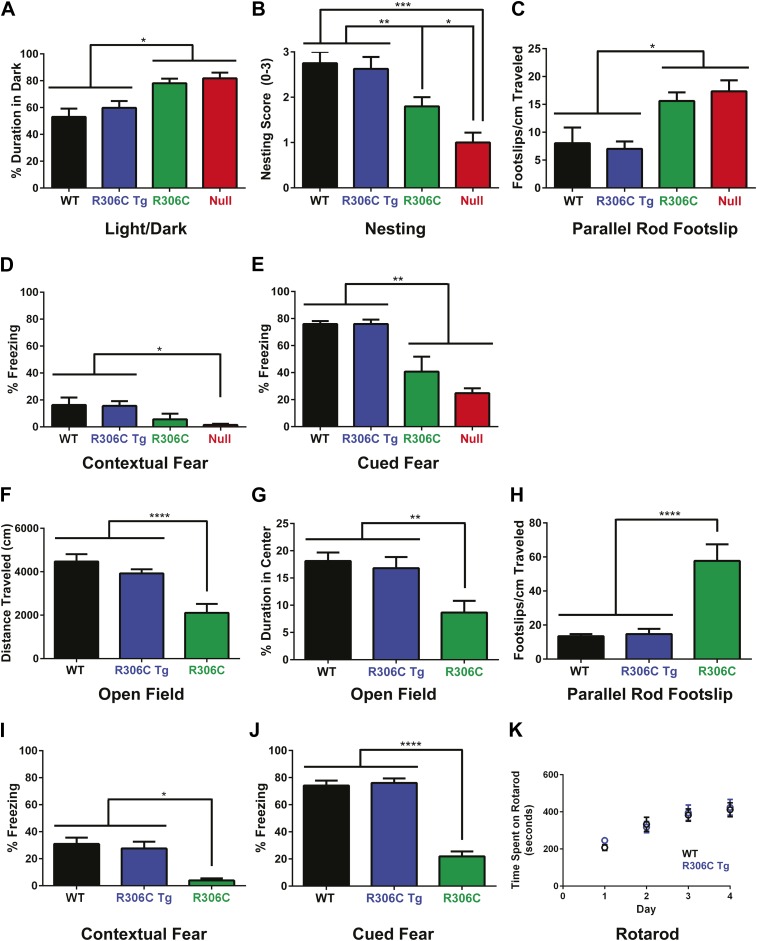
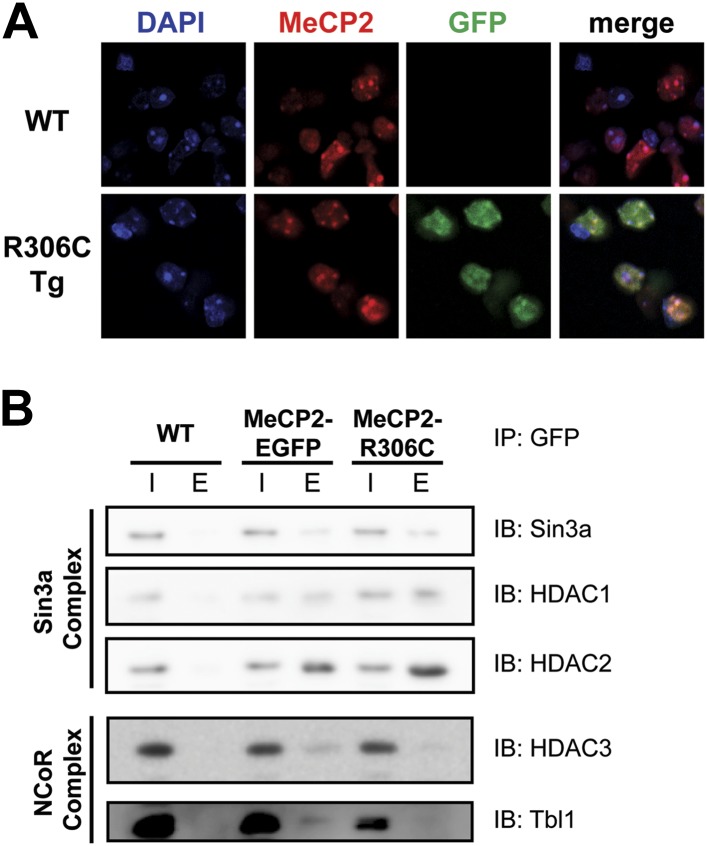
Loss of function of the X-linked gene encoding methyl-CpG binding protein 2 (MeCP2) causes the progressive neurological disorder Rett syndrome (RTT). Conversely, duplication or triplication of Xq28 causes an equally wide-ranging progressive neurological disorder, MECP2 duplication syndrome, whose features overlap somewhat with RTT. To understand which MeCP2 functions cause toxicity in the duplication syndrome, we generated mouse models expressing endogenous Mecp2 along with a RTT-causing mutation in either the methyl-CpG binding domain (MBD) or the transcriptional repression domain (TRD). We determined that both the MBD and TRD must function for doubling MeCP2 to be toxic. Mutating the MBD reproduces the null phenotype and expressing the TRD mutant produces milder RTT phenotypes, yet both mutations are harmless when expressed with endogenous Mecp2. Surprisingly, mutating the TRD is more detrimental than deleting the entire C-terminus, indicating a dominant-negative effect on MeCP2 function, likely due to the disruption of a basic cluster.
编码甲基化CpG结合蛋白2(MeCP2)的X连锁基因功能丧失会导致进行性神经疾病雷特综合征(RTT)。相反,Xq28的重复或三倍体导致同样广泛的进行性神经疾病,即MECP2重复综合征,其特征与RTT有一定重叠。为了了解MeCP2的哪些功能在重复综合征中导致毒性,我们构建了表达内源性Mecp2以及甲基化CpG结合域(MBD)或转录抑制域(TRD)中导致RTT的突变的小鼠模型。我们确定,MBD和TRD必须发挥功能,MeCP2加倍才会产生毒性。突变MBD会重现无效表型,而表达TRD突变体则会产生较轻的RTT表型,但当与内源性Mecp2一起表达时,这两种突变都是无害的。令人惊讶的是,突变TRD比删除整个C末端更有害,这表明对MeCP2功能有显性负效应,可能是由于一个碱性簇的破坏。