Ben-Shachar Shay, Chahrour Maria, Thaller Christina, Shaw Chad A, Zoghbi Huda Y
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Hum Mol Genet. 2009 Jul 1;18(13):2431-42. doi: 10.1093/hmg/ddp181. Epub 2009 Apr 15.
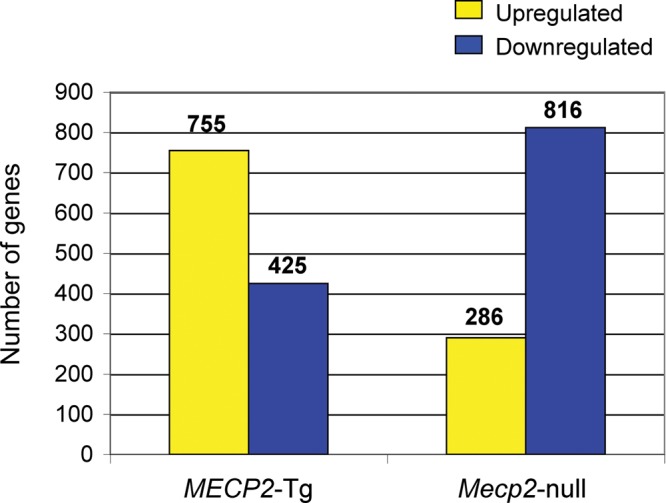
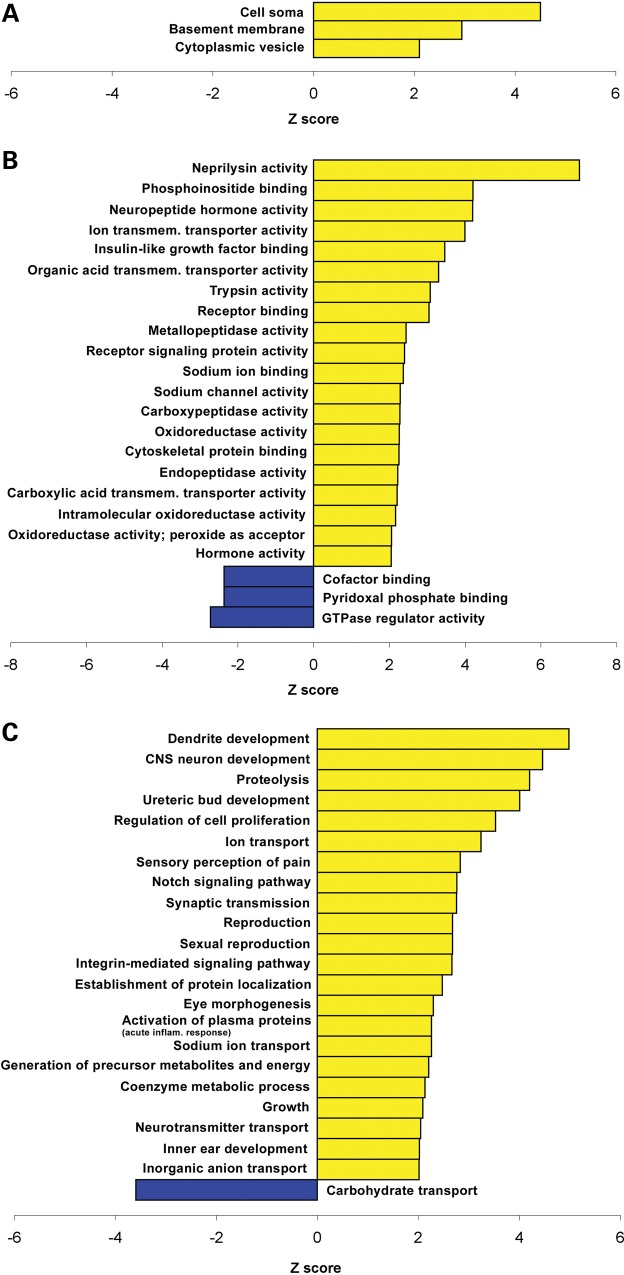
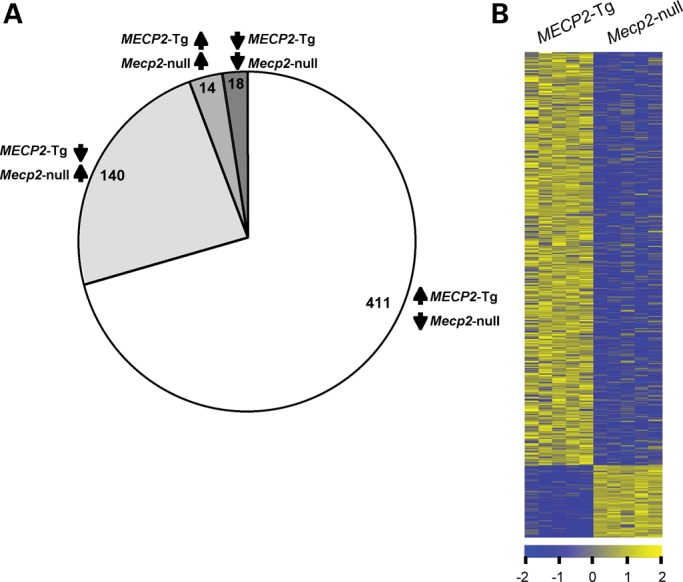
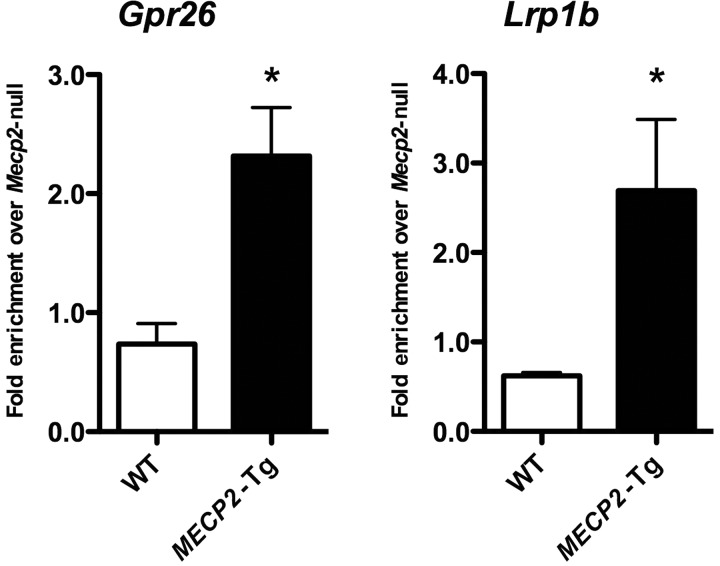
A group of post-natal neurodevelopmental disorders collectively referred to as MeCP2 disorders are caused by aberrations in the gene encoding methyl-CpG-binding protein 2 (MECP2). Loss of MeCP2 function causes Rett syndrome (RTT), whereas increased copy number of the gene causes MECP2 duplication or triplication syndromes. MeCP2 acts as a transcriptional repressor, however the gene expression changes observed in the hypothalamus of MeCP2 disorder mouse models suggest that MeCP2 can also upregulate gene expression, given that the majority of genes are downregulated upon loss of MeCP2 and upregulated in its presence. To determine if this dual role of MeCP2 extends beyond the hypothalamus, we studied gene expression patterns in the cerebellum of Mecp2-null and MECP2-Tg mice, modeling RTT and MECP2 duplication syndrome, respectively. We found that abnormal MeCP2 dosage causes alterations in the expression of hundreds of genes in the cerebellum. The majority of genes were upregulated in MECP2-Tg mice and downregulated in Mecp2-null mice, consistent with a role for MeCP2 as a modulator that can both increase and decrease gene expression. Interestingly, many of the genes altered in the cerebellum, particularly those increased by the presence of MeCP2 and decreased in its absence, were similarly altered in the hypothalamus. Our data suggest that either gain or loss of MeCP2 results in gene expression changes in multiple brain regions and that some of these changes are global. Further delineation of the expression pattern of MeCP2 target genes throughout the brain might identify subsets of genes that are more amenable to manipulation, and can thus be used to modulate some of the disease phenotypes.
一组统称为MeCP2障碍的产后神经发育障碍是由编码甲基CpG结合蛋白2(MECP2)的基因异常引起的。MeCP2功能丧失会导致雷特综合征(RTT),而该基因拷贝数增加则会导致MECP2重复或三倍体综合征。MeCP2作为一种转录抑制因子,然而在MeCP2障碍小鼠模型的下丘脑中观察到的基因表达变化表明,MeCP2也可以上调基因表达,因为大多数基因在MeCP2缺失时下调,而在其存在时上调。为了确定MeCP2的这种双重作用是否超出下丘脑,我们研究了分别模拟RTT和MECP2重复综合征的Mecp2基因敲除小鼠和MECP2转基因小鼠小脑的基因表达模式。我们发现,MeCP2剂量异常会导致小脑中数百个基因的表达发生改变。大多数基因在MECP2转基因小鼠中上调,而在Mecp2基因敲除小鼠中下调,这与MeCP2作为一种既能增加又能减少基因表达的调节因子的作用一致。有趣的是,小脑中改变的许多基因,特别是那些在MeCP2存在时增加而在其缺失时减少的基因,在下丘脑中也有类似的改变。我们的数据表明,MeCP2的增加或减少都会导致多个脑区的基因表达变化,而且其中一些变化是全局性的。进一步描绘MeCP2靶基因在全脑的表达模式可能会识别出更易于操纵的基因子集,从而可用于调节某些疾病表型。