Mottarella Scott E, Beglov Dmitri, Beglova Natalia, Nugent Matthew A, Kozakov Dima, Vajda Sandor
Program in Bioinformatics and ‡Department of Biomedical Engineering, Boston University , 44 Cummington Street, Boston, Massachusetts 02215, United States.
J Chem Inf Model. 2014 Jul 28;54(7):2068-78. doi: 10.1021/ci500115j. Epub 2014 Jul 10.
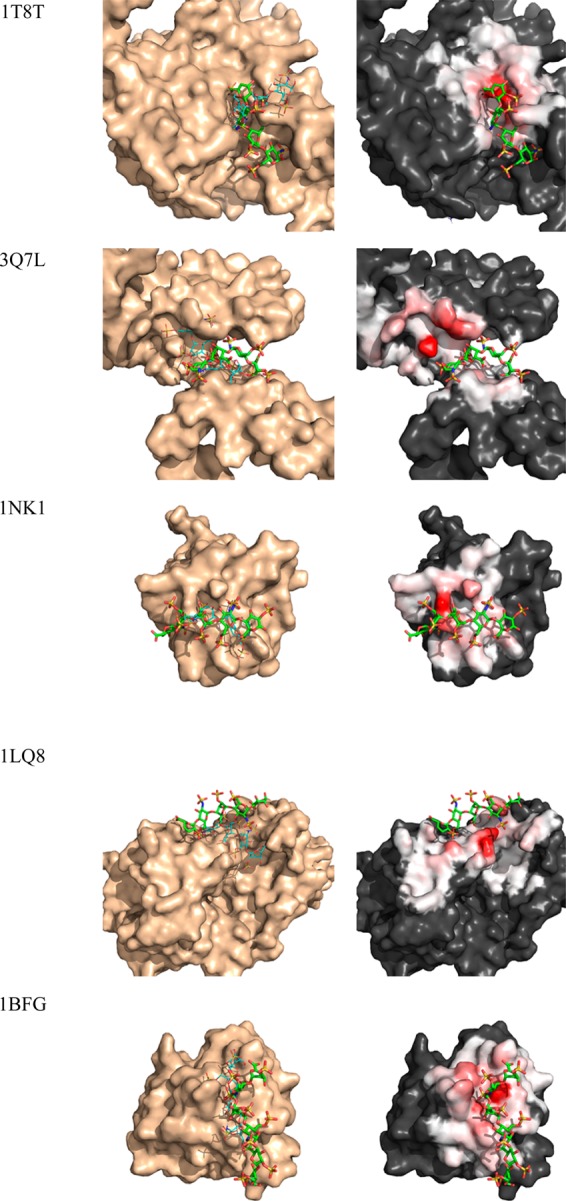
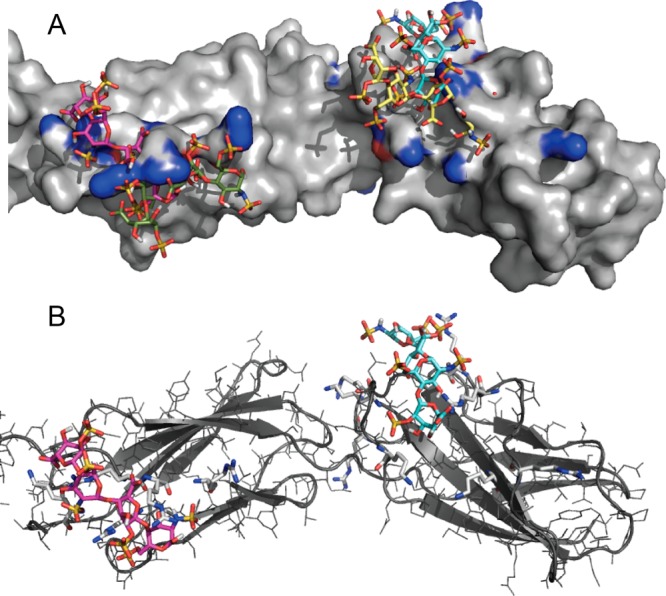
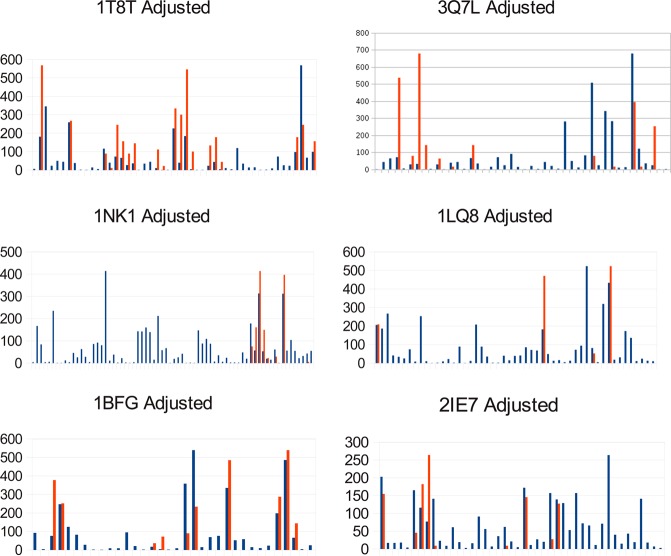
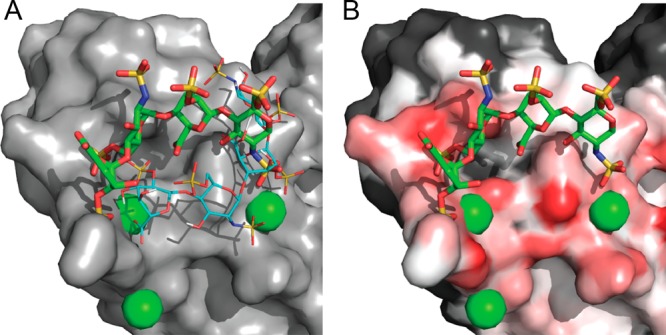
Many proteins of widely differing functionality and structure are capable of binding heparin and heparan sulfate. Since crystallizing protein-heparin complexes for structure determination is generally difficult, computational docking can be a useful approach for understanding specific interactions. Previous studies used programs originally developed for docking small molecules to well-defined pockets, rather than for docking polysaccharides to highly charged shallow crevices that usually bind heparin. We have extended the program PIPER and the automated protein-protein docking server ClusPro to heparin docking. Using a molecular mechanics energy function for scoring and the fast Fourier transform correlation approach, the method generates and evaluates close to a billion poses of a heparin tetrasaccharide probe. The docked structures are clustered using pairwise root-mean-square deviations as the distance measure. It was shown that clustering of heparin molecules close to each other but having different orientations and selecting the clusters with the highest protein-ligand contacts reliably predicts the heparin binding site. In addition, the centers of the five most populated clusters include structures close to the native orientation of the heparin. These structures can provide starting points for further refinement by methods that account for flexibility such as molecular dynamics. The heparin docking method is available as an advanced option of the ClusPro server at http://cluspro.bu.edu/ .
许多功能和结构差异很大的蛋白质都能够结合肝素和硫酸乙酰肝素。由于结晶蛋白质 - 肝素复合物以进行结构测定通常很困难,计算对接可能是理解特定相互作用的一种有用方法。以前的研究使用最初为将小分子对接至明确口袋而开发的程序,而非将多糖对接至通常结合肝素的高电荷浅裂缝的程序。我们已将PIPER程序和自动蛋白质 - 蛋白质对接服务器ClusPro扩展用于肝素对接。该方法使用分子力学能量函数进行评分,并采用快速傅里叶变换相关方法,生成并评估近十亿个肝素四糖探针的构象。对接结构使用成对均方根偏差作为距离度量进行聚类。结果表明,彼此靠近但取向不同的肝素分子聚类,并选择蛋白质 - 配体接触最多的聚类能够可靠地预测肝素结合位点。此外,五个最密集聚类的中心包含接近肝素天然取向的结构。这些结构可为通过诸如分子动力学等考虑灵活性的方法进行进一步优化提供起点。肝素对接方法可作为ClusPro服务器的高级选项在http://cluspro.bu.edu/ 上获取。