Zhang Wei, Song Wang, Zhang Zhengqing, Wang Haidong, Yang Miaomiao, Guo Ruijian, Li Menglou
College of Forestry, Northwest A&F University, Yangling, Shaanxi, People's Republic of China.
Laboratory of Agrozoology, Department of Crop Protection, Faculty of Bioscience Engineering, Ghent University, Ghent, Belgium.
PLoS One. 2014 Jun 30;9(6):e100673. doi: 10.1371/journal.pone.0100673. eCollection 2014.
Dastarcus helophoroides is known as the most valuable natural enemy insect against many large-body longhorned beetles. The molecular mechanism of its long lifespan and reproduction makes it a unique resource for genomic research. However, molecular biological studies on this parasitic beetle are scarce, and genomic information for D. helophoroides is not currently available. Thus, transcriptome information for this species is an important resource that is required for a better understanding of the molecular mechanisms of D. helophoroides. In this study, we obtained transcriptome information of D. helophoroides using high-throughput RNA sequencing.
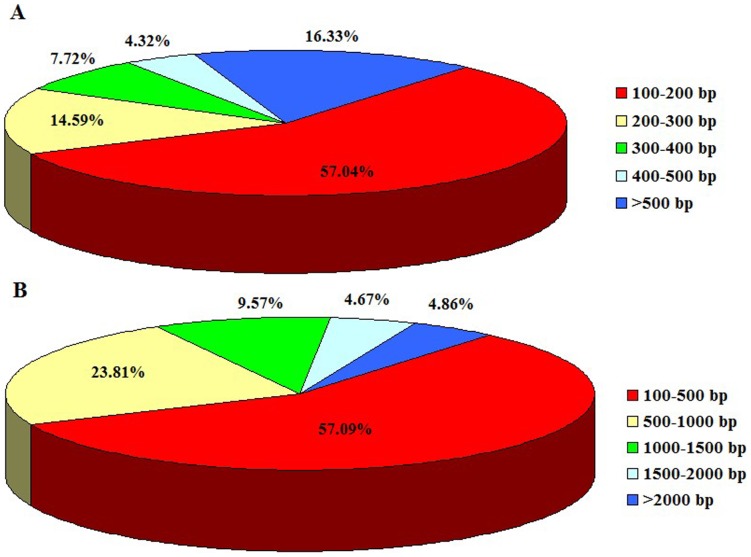
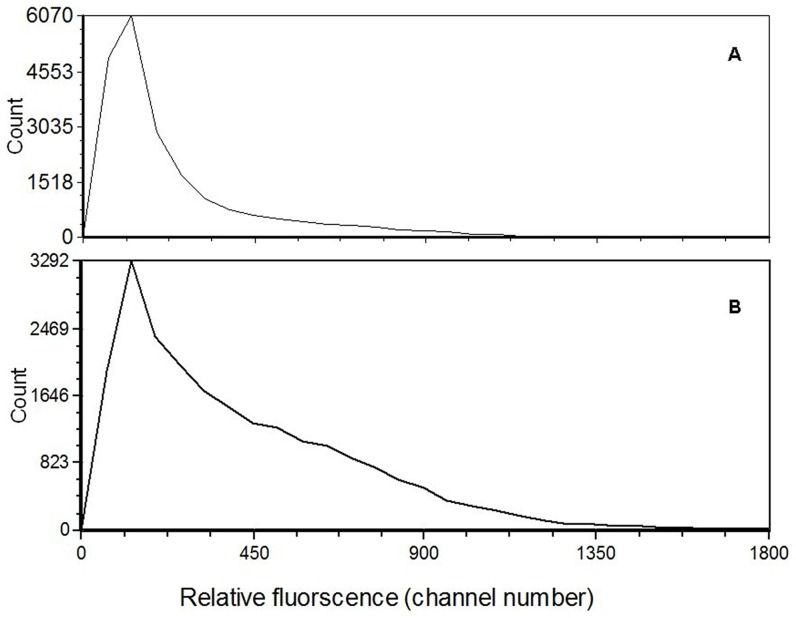
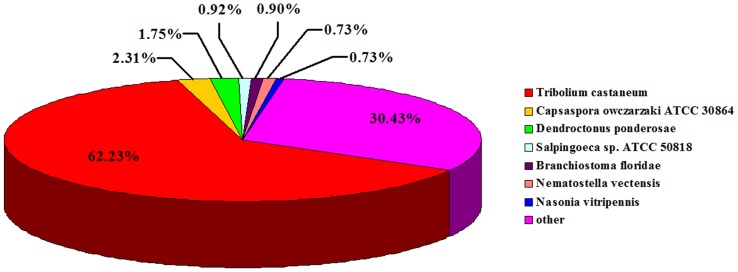
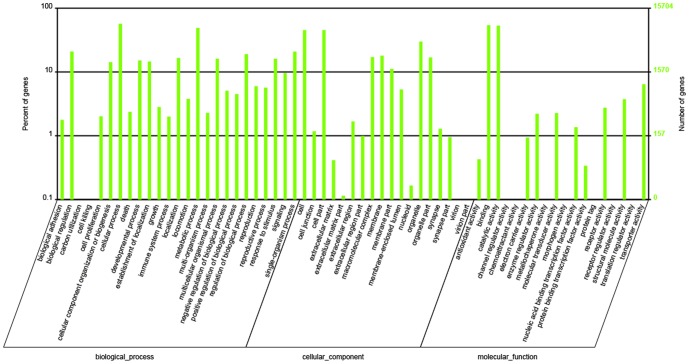
Using Illumina HiSeq 2000 sequencing, 27,543,746 clean reads corresponding to a total of 2.48 Gb nucleotides were obtained from a single run. These reads were assembled into 42,810 unigenes with a mean length of 683 bp. Using a sequence similarity search against the five public databases (NR, Swiss-Prot, GO, COG, KEGG) with a cut-off E-value of 10(-5) using Blastx, a total of 31,293 unigenes were annotated with gene description, gene ontology terms, or metabolic pathways.
To the best of our knowledge, this is the first study on the transcriptome information of D. helophoroides. The transcriptome data presented in this study provide comprehensive information for future studies in D. helophoroides, particularly for functional genomic studies in this parasitic beetle.
花绒寄甲是已知的针对多种大型天牛最具价值的天敌昆虫。其长寿和繁殖的分子机制使其成为基因组研究的独特资源。然而,关于这种寄生甲虫的分子生物学研究较少,目前尚无花绒寄甲的基因组信息。因此,该物种的转录组信息是更好地理解花绒寄甲分子机制所需的重要资源。在本研究中,我们使用高通量RNA测序获得了花绒寄甲的转录组信息。
使用Illumina HiSeq 2000测序,单次运行获得了27,543,746条clean reads,对应总共2.48 Gb核苷酸。这些reads被组装成42,810个单基因,平均长度为683 bp。使用Blastx对五个公共数据库(NR、Swiss-Prot、GO、COG、KEGG)进行序列相似性搜索,截止E值为10(-5),共有31,293个单基因被注释了基因描述、基因本体术语或代谢途径。
据我们所知,这是首次对花绒寄甲转录组信息进行的研究。本研究中呈现的转录组数据为花绒寄甲未来的研究提供了全面信息,特别是针对这种寄生甲虫的功能基因组学研究。