Luczak Magdalena, Marczak Lukasz, Stobiecki Maciej
Institute of Bioorganic Chemistry, Polish Academy of Sciences, Poznan, Poland.
PLoS One. 2014 Jul 2;9(7):e101694. doi: 10.1371/journal.pone.0101694. eCollection 2014.
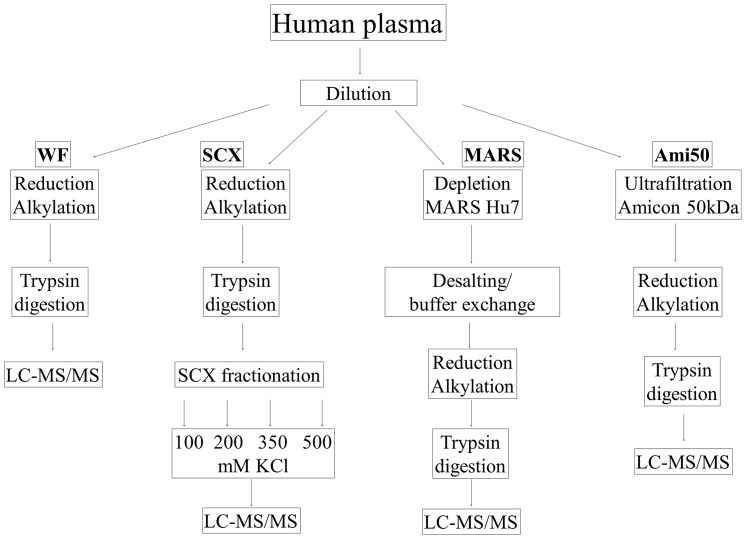
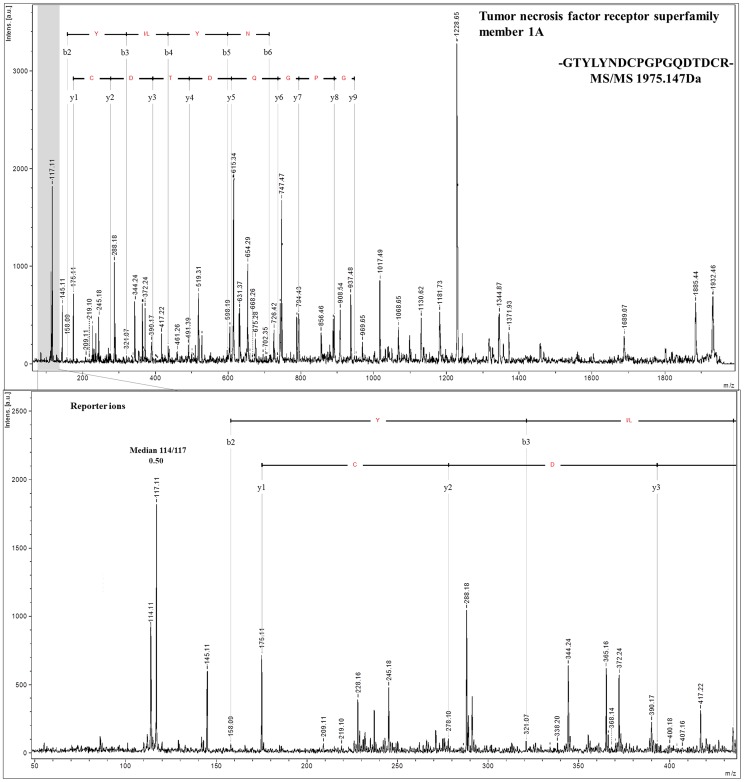
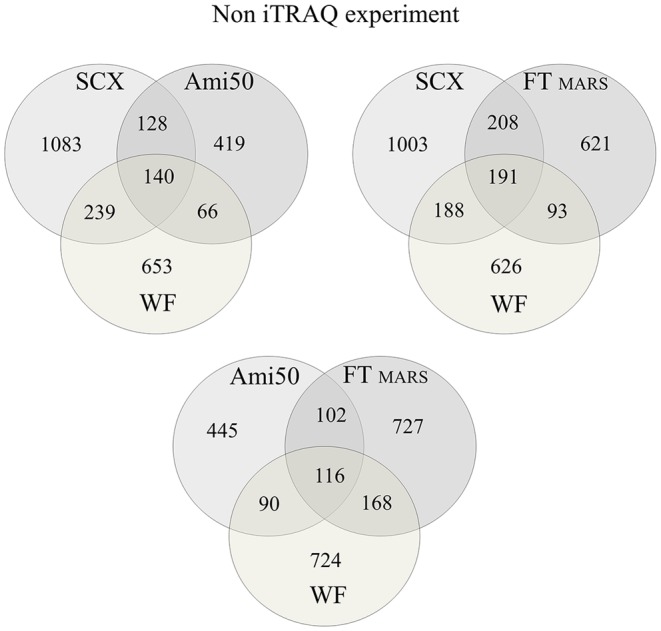
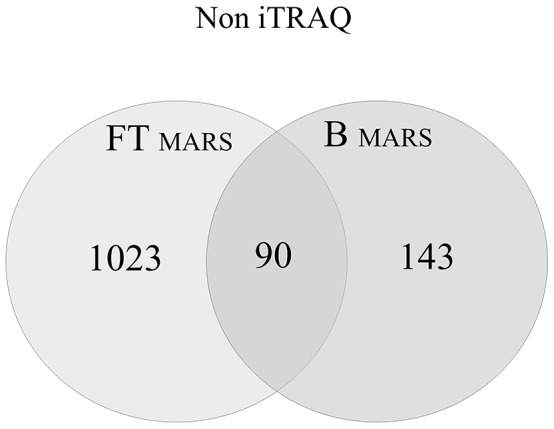
Shotgun proteomic methods involving iTRAQ (isobaric tags for relative and absolute quantitation) peptide labeling facilitate quantitative analyses of proteomes and searches for useful biomarkers. However, the plasma proteome's complexity and the highly dynamic plasma protein concentration range limit the ability of conventional approaches to analyze and identify a large number of proteins, including useful biomarkers. The goal of this paper is to elucidate the best approach for plasma sample pretreatment for MS- and iTRAQ-based analyses. Here, we systematically compared four approaches, which include centrifugal ultrafiltration, SCX chromatography with fractionation, affinity depletion, and plasma without fractionation, to reduce plasma sample complexity. We generated an optimized protocol for quantitative protein analysis using iTRAQ reagents and an UltrafleXtreme (Bruker Daltonics) MALDI TOF/TOF mass spectrometer. Moreover, we used a simple, rapid, efficient, but inexpensive sample pretreatment technique that generated an optimal opportunity for biomarker discovery. We discuss the results from the four sample pretreatment approaches and conclude that SCX chromatography without affinity depletion is the best plasma sample preparation pretreatment method for proteome analysis. Using this technique, we identified 1,780 unique proteins, including 1,427 that were quantified by iTRAQ with high reproducibility and accuracy.
涉及iTRAQ(相对和绝对定量的等压标签)肽标记的鸟枪法蛋白质组学方法有助于蛋白质组的定量分析和寻找有用的生物标志物。然而,血浆蛋白质组的复杂性以及血浆蛋白浓度的高度动态范围限制了传统方法分析和鉴定大量蛋白质(包括有用的生物标志物)的能力。本文的目的是阐明基于质谱和iTRAQ分析的血浆样品预处理的最佳方法。在这里,我们系统地比较了四种方法,包括离心超滤、分级分离的强阳离子交换色谱、亲和去除和未分级的血浆,以降低血浆样品的复杂性。我们使用iTRAQ试剂和UltrafleXtreme(布鲁克道尔顿公司)基质辅助激光解吸电离飞行时间/串联飞行时间质谱仪生成了定量蛋白质分析的优化方案。此外,我们使用了一种简单、快速、高效但廉价的样品预处理技术,为生物标志物发现创造了最佳机会。我们讨论了四种样品预处理方法的结果,并得出结论,无亲和去除的强阳离子交换色谱是蛋白质组分析中最佳的血浆样品制备预处理方法。使用该技术,我们鉴定了1780种独特的蛋白质,其中1427种通过iTRAQ进行了高度可重复和准确的定量。