van Stipdonk Michael J, Basu Partha, Dille Sara A, Gibson John K, Berden Giel, Oomens Jos
Department of Chemistry and Biochemistry, Duquesne University , 600 Forbes Avenue, Pittsburgh, Pennsylvania 15282, United States.
J Phys Chem A. 2014 Jul 24;118(29):5407-18. doi: 10.1021/jp503222v. Epub 2014 Jul 10.
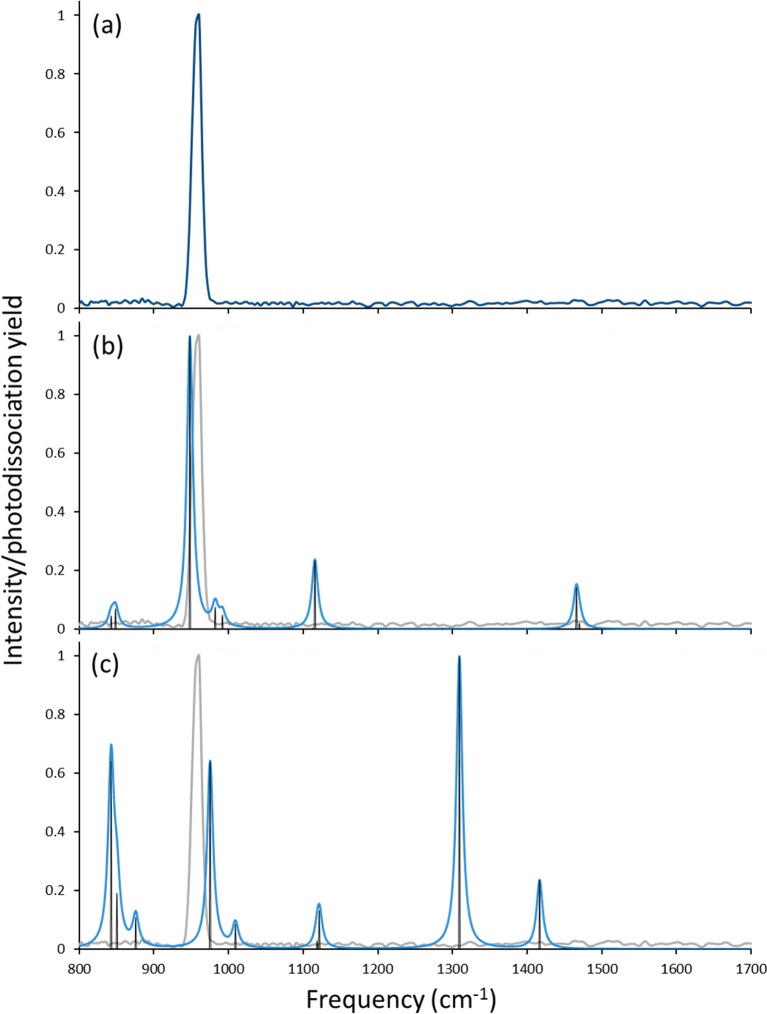
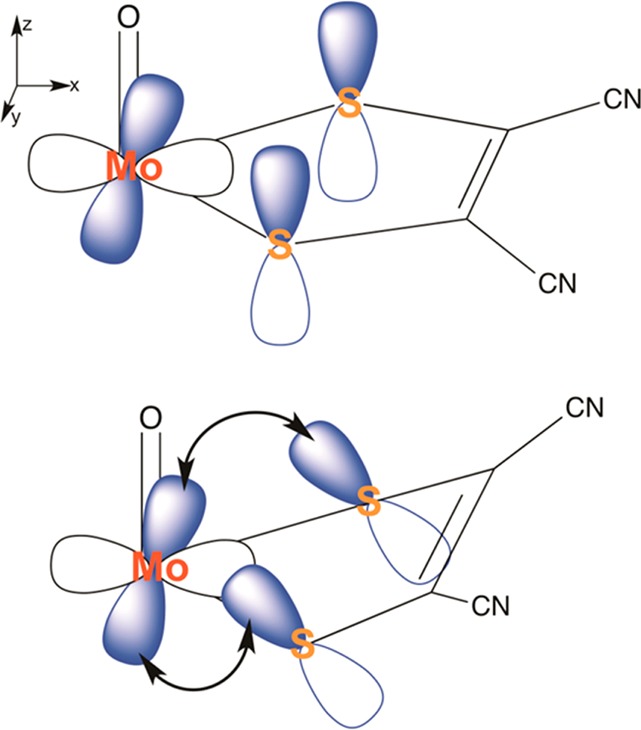
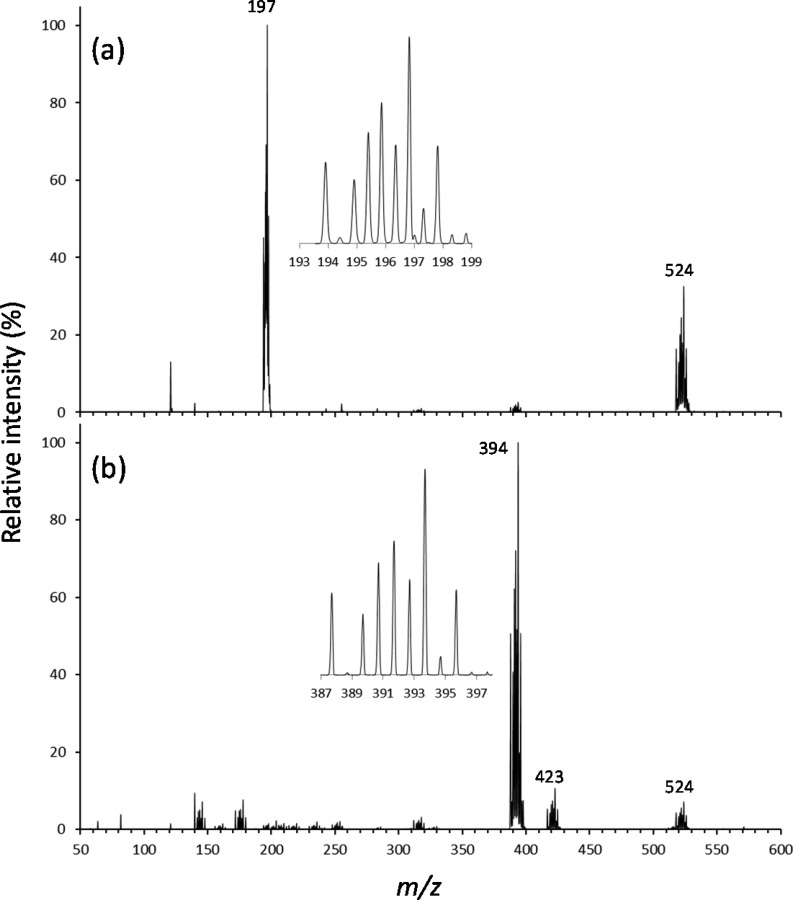
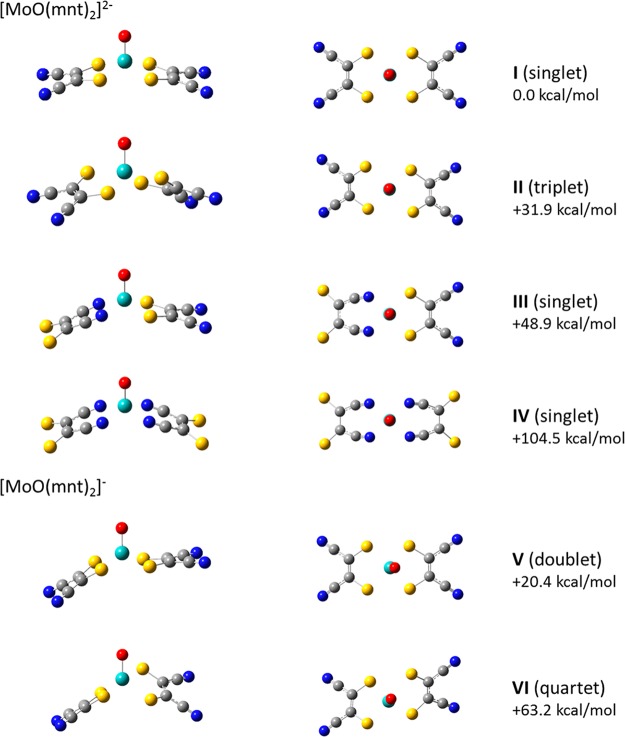
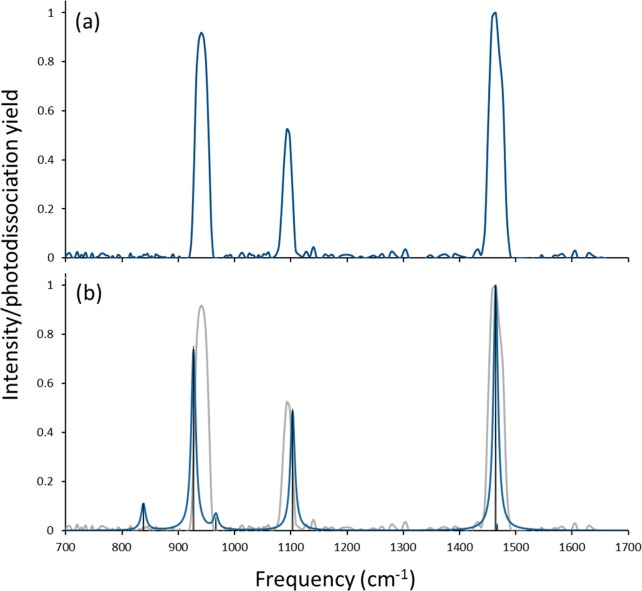
Electrospray ionization (ESI) in the negative ion mode was used to create anionic, gas-phase oxo-molybdenum complexes with dithiolene ligands. By varying ESI and ion transfer conditions, both doubly and singly charged forms of the complex, with identical formulas, could be observed. Collision-induced dissociation (CID) of the dianion generated exclusively the monoanion, while fragmentation of the monoanion involved decomposition of the dithiolene ligands. The intrinsic structure of the monoanion and the dianion were determined by using wavelength-selective infrared multiple-photon dissociation (IRMPD) spectroscopy and density functional theory calculations. The IRMPD spectrum for the dianion exhibits absorptions that can be assigned to (ligand) C ═ C, C-S, C-C ≡ N, and Mo ═ O stretches. Comparison of the IRMPD spectrum to spectra predicted for various possible conformations allows assignment of a pseudo square pyramidal structure with C2v symmetry, equatorial coordination of MoO(2+) by the S atoms of the dithiolene ligands, and a singlet spin state. A single absorption was observed for the oxidized complex. When the same scaling factor employed for the dianion is used for the oxidized version, theoretical spectra suggest that the absorption is the Mo ═ O stretch for a distorted square pyramidal structure and doublet spin state. A predicted change in conformation upon oxidation of the dianion is consistent with a proposed bonding scheme for the bent-metallocene dithiolene compounds [Lauher, J. W.; Hoffmann, R. J. Am. Chem. Soc. 1976 , 98 , 1729 - 1742], where a large folding of the dithiolene moiety along the S · · · S vector is dependent on the occupancy of the in-plane metal d-orbital.
在负离子模式下,采用电喷雾电离(ESI)来制备带有二硫烯配体的阴离子气相氧代钼配合物。通过改变ESI和离子转移条件,可以观察到具有相同分子式的配合物的双电荷和单电荷形式。二价阴离子的碰撞诱导解离(CID)仅产生一价阴离子,而一价阴离子的碎片化涉及二硫烯配体的分解。利用波长选择性红外多光子解离(IRMPD)光谱和密度泛函理论计算确定了一价阴离子和二价阴离子的内在结构。二价阴离子的IRMPD光谱显示出可归属于(配体)C═C、C-S、C-C≡N和Mo═O伸缩振动的吸收峰。将IRMPD光谱与各种可能构象预测的光谱进行比较,确定其为具有C2v对称性的假四方锥结构,二硫烯配体的S原子在赤道平面配位MoO(2+),且为单重态自旋状态。氧化态配合物观察到单一吸收峰。当对二价阴离子使用的相同缩放因子用于氧化态版本时,理论光谱表明该吸收峰是扭曲四方锥结构和双重态自旋状态的Mo═O伸缩振动。二价阴离子氧化时预测的构象变化与弯曲金属茂二硫烯化合物提出的键合方案一致[Lauher, J. W.; Hoffmann, R. J. Am. Chem. Soc. 1976, 98, 1729 - 1742],其中二硫烯部分沿S···S向量的大幅折叠取决于面内金属d轨道的占据情况。